
气相色谱-质谱联用法测定含植物提取物类化妆品中滴滴伊的不确定度评定
2022-12-28 07:53:40严敏彭娟顾娟红
化学分析计量 2022年11期
严敏,彭娟,顾娟红
(1.苏州世标检测技术有限公司,江苏苏州 215104; 2.苏州海关综合技术中心,江苏苏州 215104)
测量不确定度是表征合理地赋予被测量值的分散性、与测量结果相联系的参数,测量过程中的随机效应和系统效应均会导致测量不确定度[1-2],对检测方法进行不确定度评估既是国家计量技术规范中的规定,也是确定测量结果科学、有效性的保证[3-5]。
笔者按照GB/T 39665—2020 《含植物提取物类化妆品中55 种禁用农药残留量的测定》规定,用乙腈涡旋振荡、超声提取乳液样品,提取液经凝胶色谱仪净化后,采用气相色谱-质谱法进行测定,外标法定量。参考测量不确定度评定与表示的相关标准与文献[1,6-15],对含植物提取物类化妆品中禁用的两种滴滴伊残留量检测结果进行不确定度评定,建立了数学模型,分析了不确定度产生的原因,给出了分析结果的置信区间,对评判检测结果的可比性和可接受性具有重要意义。
1 实验部分
1.1 主要仪器与试剂
气相色谱-质谱联用仪:8890+5977B 型,美国安捷伦科技有限公司。
凝 胶 渗 透 色 谱 仪:FREESTYLE 型,德 国LCTech 公司。
旋转蒸发仪:R-215 型,瑞士步琦有限公司。
Milli-Q 纯水机:美国密理博公司。
分析天平:Mettler EME 204 型,梅特勒-托利多仪器有限公司。
旋涡仪:MS3 型,德国IKA 有限公司。
氮吹仪:NEVP-12 型,江苏天瑞仪器股份有限公司。
离心机:TDZ5-WS 型,上海卢湘仪离心机仪器有限公司。
超声波清洗器:KH-500DE 型,昆山禾创超声仪器有限公司。
有机相滤膜:孔径为0.45 μm。
滴滴伊混合标准样品:含有o,p'-DDE 、p,p'-DDE,二者质量浓度均为50.0 μg/mL,溶剂为丙酮,天津阿尔塔科技有限公司。
丙酮、环己烷、乙酸乙酯、乙腈、正己烷:色谱纯,上海安谱实验科技股份有限公司。
NaCl:分析纯,苏州国药集团,使用前于(140±10) ℃烘烤4 h,置于干燥器中冷却,备用。
空白乳液样品:乳液样品,按照标准方法检测不含目标物。
氦气:纯度为99.999%。实验用水为一级水。
1.2 样品提取
准确称取乳液样品1 g(精确至0.001 g),置于50 mL 具塞离心管中,加入10 mL 乙腈,涡旋振荡1 min,超声提取10 min,加入5~7 g 氯化钠,涡旋1 min 后,以8000 r/min 转速离心2 min。将上层清液移入新的50 mL 具塞离心管中,残渣再用10 mL 乙腈重复提取一次,上层清液合并于同一离心管中,在(40±2)℃水浴温度下,用氮气浓缩装置浓缩至近干,待净化。
1.3 样品净化
1.3.1 洗脱条件
凝胶渗透色谱柱:400 mm×25 mm,内装聚苯乙烯凝胶填料(粒径为38~80 μm);洗脱溶剂:环己烷-乙酸乙酯溶液(体积比为1∶1,下同);预洗脱时间:15 min;采集时间:15 min;柱冲洗时间:10 min;进样体积:5 mL;流量:5 mL/min。
1.3.2 净化
向离心管中加入10 mL 环己烷-乙酸乙酯溶液溶解残留物,然后转移至10 mL 容量瓶中并定容,经有机相滤膜过滤于凝胶渗透色谱仪进样瓶中。按1.3.1 条件净化后,转移收集液于250 mL 平底烧瓶中,凝胶渗透色谱仪收集瓶用10~20 mL 环己烷-乙酸乙酯溶液清洗2 次,合并至250 mL 平底烧瓶中,在(40±2) ℃水浴温度下,用旋转蒸发仪蒸发近干。准确加入0.5 mL 正己烷溶解残渣,混匀,经有机相滤膜过滤,供气相色谱-质谱仪测定。
1.4 标准溶液配制
滴滴伊混合标准溶液:两种滴滴伊质量浓度均为500 μg/L,准确移取0.1 mL 滴滴伊混合标准样品于10 mL 容量瓶中,用丙酮稀释并定容。
滴滴伊系列混合标准工作溶液:准确移取一定量滴滴伊混合标准溶液,用丙酮稀释,得到质量浓度均分别为20.0、50.0、100、200、500 μg/L 的系列混合标准工作溶液。
空白乳液样品基质溶液:分别称取1 g(精确至0.001 g)乳液空白样品,按照1.2 和1.3 处理至“合并至250 mL 平底烧瓶中”,得空白乳液样品基质溶液。
基质混合标准工作溶液:用氮气将空白样品基质溶液吹干,准确加入0.5 mL 标准工作溶液复溶,经有机相滤膜过滤,得到基质混合标准工作溶液,待测。
1.5 仪器工作条件
1.5.1 气相色谱仪
色谱柱:HP-5MS UI 毛细管柱[30 m×0.250 mm,0.25 μm,安捷伦科技(中国)有限公司];进样口温度:260 ℃;程序升温:初始温度为70 ℃,保持2 min,以25 ℃/min 升至150 ℃,再以3 ℃/min升至280 ℃,保持10 min;载气:氦气,流量为1.0 mL/min ;进样方式:不分流进样;进样体积:1 μL。
1.5.2 质谱仪
电离方式:EI 电离源;电离能:70 eV;离子源温度:230 ℃;四级杆温度:150 ℃;接口温度:280℃;扫描方式:选择离子扫描模式(SIM);溶剂延迟:4 min;定性方法:色谱峰保留时间及特征碎片质荷比;定量方法:色谱峰面积外标法定量;定性离子和定量离子信息见表1。

表1 两种滴滴伊定性及定量离子
2 数学模型
样品中农药残留量按式(1)计算:
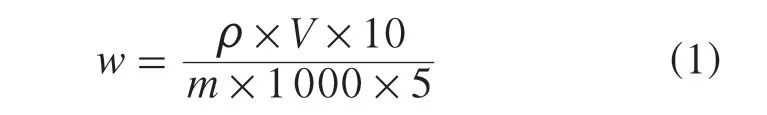
式中:w——样品中待测组分的质量分数,mg/kg;
ρ——从标准曲线得到待测组分的质量浓度,μg/L;
V——样品溶液最终定容体积,mL;
10——净化液定容体积,mL;
m——样品质量,g;
1 000——换算系数;
5——凝胶渗透色谱仪进样体积,mL。
从测定方法和数学模型可以看出,主要步骤包括样品称重、样品提取、GPC 净化并浓缩定容、系列标准工作溶液的制备、仪器分析等。
3 不确定度评定
3.1 测量重复性引入的相对标准不确定度ur(r)

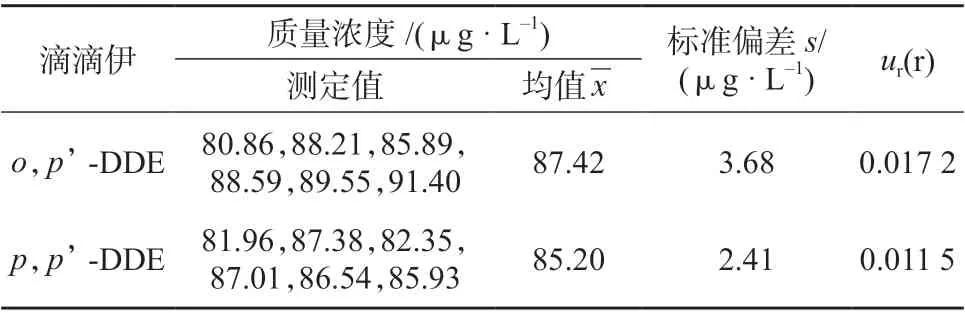
表2 重复测定结果及不确定度
3.2 样品称量引入的相对标准不确定度ur(m)
EME 204 分析天平检定证书给定的测量不确定度U=0.2 mg(k=2),则样品称量引入的标准不确定度u(m)=U/k=0.1 mg,样品6 次平行的称量均值为0.995 8 g,计算得:

3.3 待净化液定容体积引入的相对标准不确定度ur(V10)


3.4 净化后定容体积引入的相对标准不确定度ur(V )
净化后加入用1 mL 移液枪准确加入0.5 mL 正己烷,溶解残渣,由移液器校准证书可得,包含因子k=2 时,移液体积的扩展不确定度U=0.3 μL,净化后定容体积引入的标准不确定度:
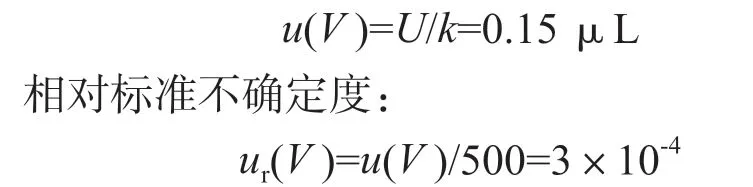
3.5 样品溶液质量浓度测定引入的相对标准不确定度分量ur(ρ)
样品溶液质量浓度测定引入的不确定度分量主要由标准储备液的不确定度、标准溶液稀释过程引入的不确定度及校准曲线拟合引入的不确定度三部分组成。
3.5.1 标准储备液的相对标准不确定度ur(P)
根据标准溶液证书,按照均匀分布计算,k= 3,由纯度(P)引入的不确定度和相对不确定度分别为u(P)=U/k,ur(P)=u(P)/P。两种滴滴伊标准品的纯度及引入的相对不确定度计算结果见表3。

表3 两种禁用农药标准品的不确定度
3.5.2 标准溶液稀释过程引入的相对标准不确定度ur(D)
在标准溶液稀释过程中使用到10 mL 容量瓶,200 μL、1 mL 和5 mL 移液枪,稀释过程见1.4。不确定度主要来源于容量瓶和移液枪的体积校准不确定度。假设是均匀分布,移液器和容量瓶的相对不确定度见表4。
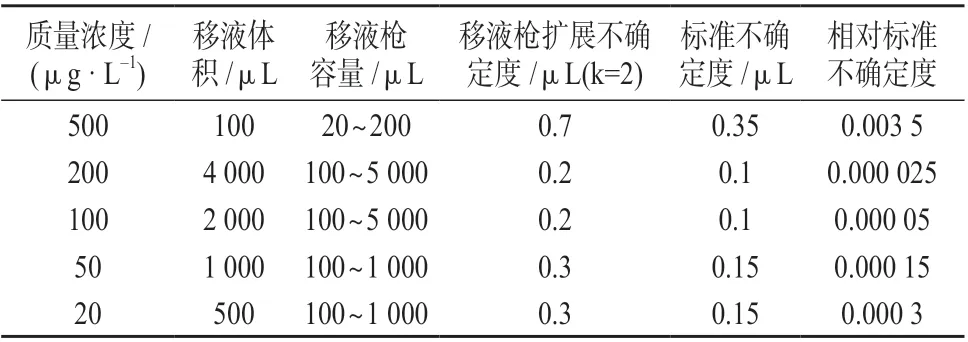
表4 两种禁用农药对照品的相对标准不确定度
对表4 中各质量浓度的相对标准不确定度进行合成,得标准工作溶液稀释过程引入的相对标准不确定度:ur(D)=0.003 516。
3.5.3 校准曲线拟合引入的相对标准不确定度ur(ρx)
对配制的标准工作溶液进行测定,每种浓度平行测定3 次,取其平均值进行线性拟合,得到标准曲线线性方程y=b+aρ及相关系数。加标样品平行测定6 次,取平均值,由拟合的标准曲线计算两种滴滴伊农药残留浓度。拟合标准偏差按式(2)计算:


标准工作曲线拟合数据及不确定度计算结果见表5。
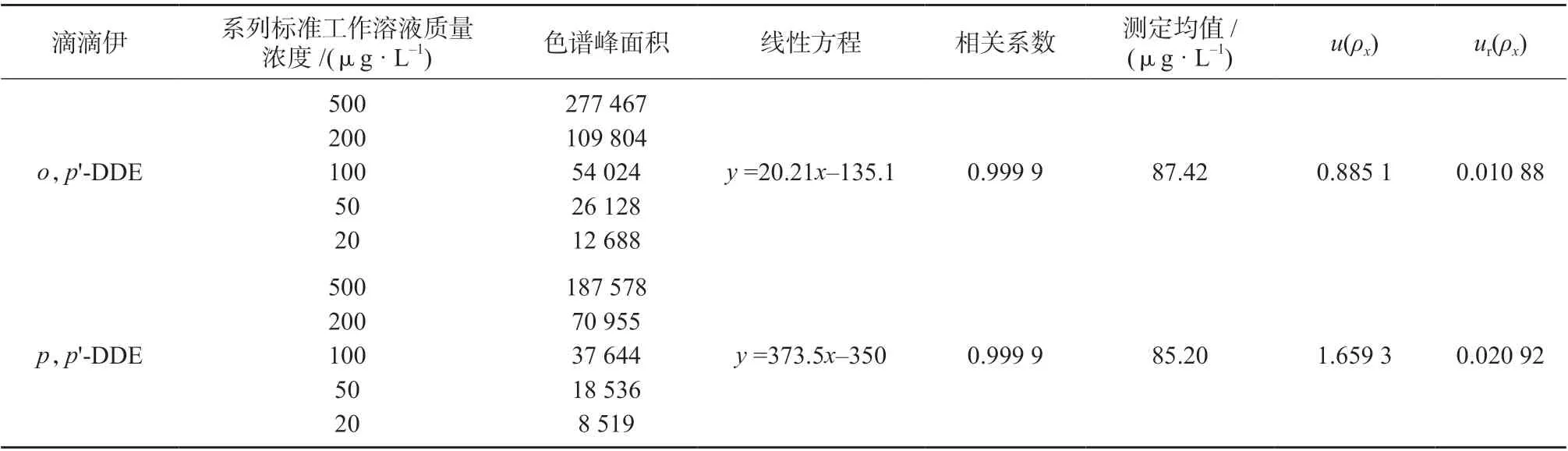
表5 标准曲线线性拟合数据及不确定度计算结果
以上不确定度分量互不相关,则样品溶液浓度测定引入的相对标准不确定度按式(5)计算:

将相关数据代入式(5),计算得o,p'-DDE、p,p'-DDE 质量浓度的相对标准不确定度分别为0.012 81、0.021 99。
3.6 气相色谱-质谱测定引入的相对标准不确定度ur(Q)
查仪器校准证书可知,气相色谱-质谱仪测量色谱峰面积的重复性相对标准偏差为2.8%,k=2,其相对标准不确定度:

3.7 合成标准不确定度和扩展不确定度
将上述各相对标准不确定度分量按照式(6)合成得测定值的相对标准不确定度,将相关测定结果代入数学模型得到样品中滴滴伊的质量分数,结果见表6。
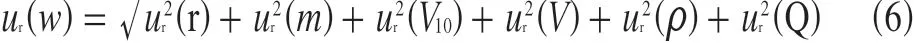

表6 合成相对标准不确定度和扩展不确定度(k=2)
3.8 不确定度报告
样品中o,p'-DDE 测定结果的质量分数为(0.087 8±0.004 5) mg/kg,k=2;p,p'-DDE 测定结果的质量分数为(0.085 6±0.004 9) mg/kg,k=2。
4 结语
采用气质联用测定含植物提取物类化妆品中两种滴滴伊残留量,在各影响测量不确定度的主要来源中,由测量重复性、样品溶液浓度测定及仪器分析所引入的不确定度影响较大,在实际检测中,应做好平行样品的测定,对仪器进行检定和维护使其保持较高灵敏度,还可以增加系列标准工作溶液浓度点数,从而降低测量不确定度。
猜你喜欢
快乐作文(1.2年级)(2021年11期)2021-12-19 00:54:04
化工设计通讯(2020年10期)2020-09-17 14:43:16
当代水产(2019年3期)2019-05-14 05:42:48
汽车观察(2019年2期)2019-03-15 06:00:00
少先队活动(2018年5期)2018-12-29 12:12:36
Coco薇(2017年7期)2017-07-21 16:49:50
Cancer Biology & Medicine(2016年4期)2017-01-13 01:54:45
中国氯碱(2016年9期)2016-11-16 03:07:39
金色年华(2016年23期)2016-06-15 20:28:28
中国科技信息(2015年24期)2015-11-07 08:52:27
