
PIK3R2基因错义变异所致MPPH综合征I型患儿遗传学分析
2022-12-16 01:43:28阮妙华胡思思周晓军石佳旻陈源朱棉棉王楸王丹
温州医科大学学报 2022年12期
阮妙华,胡思思,周晓军,石佳旻,陈源,朱棉棉,王楸,王丹
温州医科大学附属第一医院 儿科,浙江 温州 325015
巨脑畸形-多小脑回-多指(趾)畸形-脑积水(megalencephaly-polymicrogyria-polydactylyhydrocephalus, MPPH)综合征在线人类孟德尔遗传数据库(Online Mendelian Inheritance in Man,OMIM)编号6603387,是一种罕见的常染色体显性遗传病[1],分为MPPH I型、II型和III型,以脑积水、巨脑畸形、多小脑回、脑室扩大、轴后多指(趾)、癫痫发作、全面性发育迟缓伴极重度智力障碍等临床表现为特点,胎儿期最常见的表现为脑积水,新生儿期以巨脑畸形为特点,总体临床预后不良。MPPH由MIRAZAA等[1]在2004年首次提出,其中由PIK3R2基因突变引起的MPPH I型综合征,截至2021年12月31日,世界范围内可检索到16例完整临床案例报 道[2-14]。目前国内未见相关病例报道。本研究对1例智力运动发育相对落后,特殊面容和先天性脑积水的患儿进行了基因检测,探讨其遗传学病因。
1 对象和方法
1.1 对象
1.1.1 一般情况:患儿,男,出生后不久因“头围增大”就诊。患儿系第4胎第2产,出生胎龄38+5周,出生体质量3.43 kg。其母孕22周胎儿超声提示脑积水,家属拒绝行进一步产前检查。入院查体发现特殊外貌,前额突出,眼距宽,耳位低,前囟 3.5 cm×4.5 cm,平坦,头围40 cm(>3 SD),乳距宽,见图1。父母表型均正常,系非近亲结婚,家族中无类似患者。
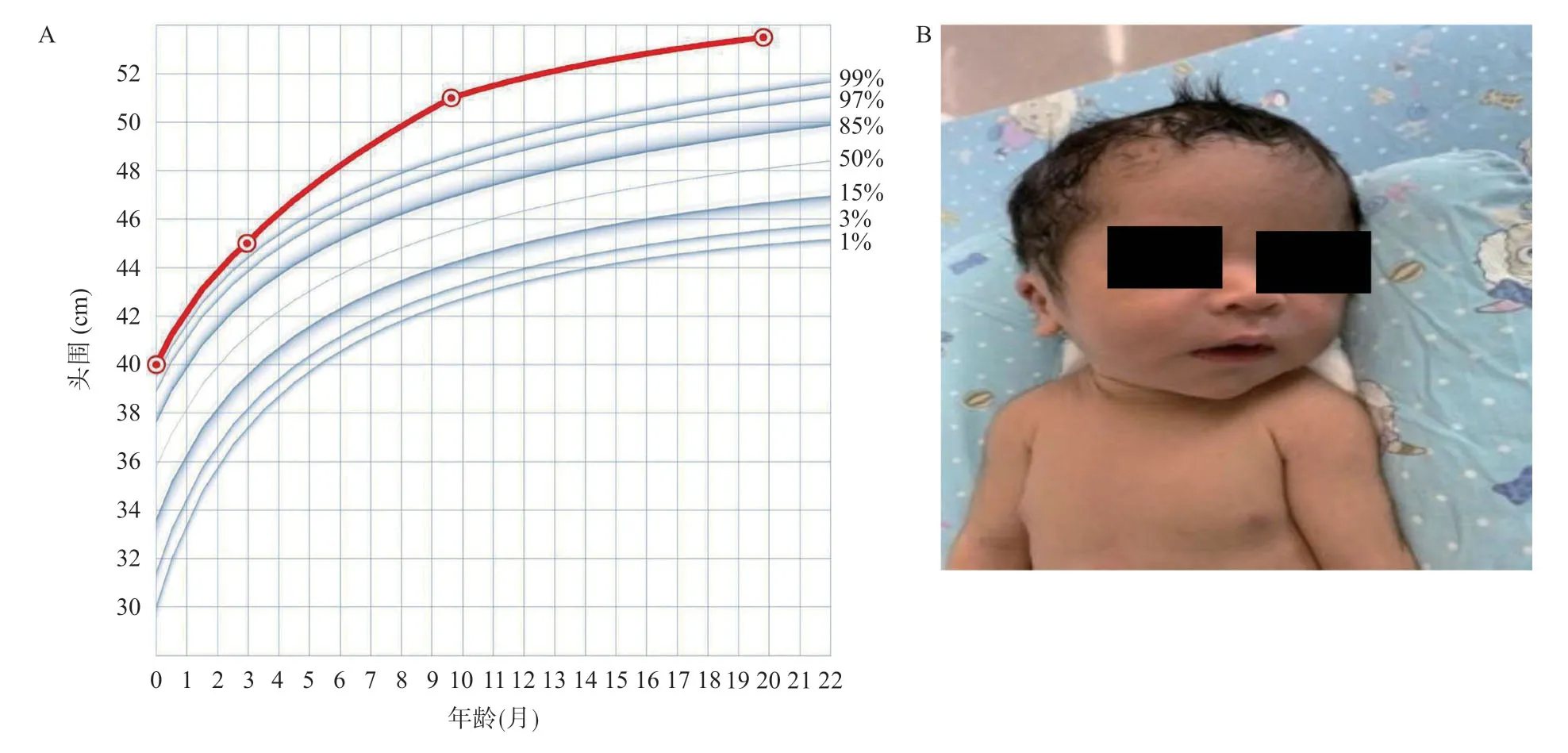
图1 先证者的临床表型
1.1.2 随访:出生后第2天头颅MRI示两侧脑室枕角少许积血伴幕上脑室轻度扩张,全脑对称性两侧额颞枕叶多小脑回畸形;脑电图检查未见明显异常。出生后19个月门诊随访:头围53.4 cm(见图1),前囟2 cm×2 cm,语言认知及运动发育相对落后,只会叫爸爸、妈妈;能独走,不会跑跳,无癫痫发作。GESELL心理学检查量表提示发育商77,提示边缘状态(76≤DQ≤85)。复查头颅MRI提示轻度幕上脑积水,渗出脑室旁脑白质髓鞘发育不良(见图2)。
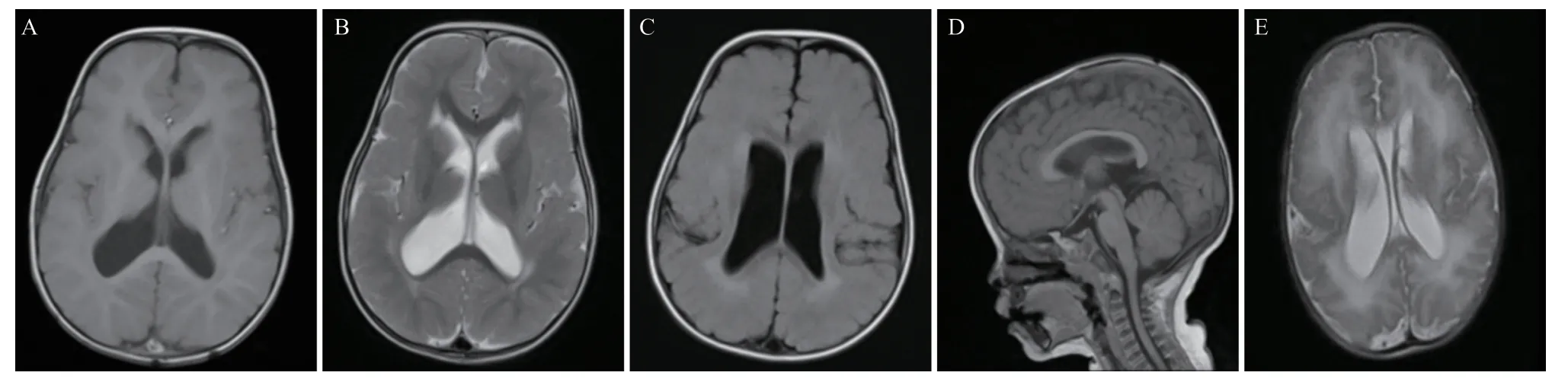
图2 头颅MRI检查结果
1.2 方法
1.2.1 基因组DNA提取:在征得其父母知情同意后,采集先证者及其父母的外周静脉血样各5 mL,用北京天根生化科技有限公司生产的试剂盒提取基因组DNA。
1.2.2 全外显子组测序和Sanger测序验证:使用IDT公司xGen®Exome Research Panel v2.0捕获探针,并用NovaSeq6000测序仪(美国Illumina公司)进行高通量测序(由北京智因东方转化医学研究中心完成)。测序目标区域覆盖度>99%,20×覆盖度达到98%,捕获效率为0.89。最后根据二代测序的结果对候选变异位点进行Sanger测序验证序,同时对其父母进行检测。
1.2.3 生物信息学分析:用Mutation Taster (http://www.mutationtaster.org/)、SIFT(http://sift/bii/a-star.edu.sg/)、PolyPhen-2、PROVEAN、CADD(https://cadd.gs.washington.edu/)等软件对变异位点的有害性进行预测。通过Clustal X-2.1将变异氨基酸与另外7个物种的氨基酸序列(来源:http://www.nci.nlm.nih.gov/homologene)进行比对,包括黑猩猩(Pan troglodytes)、家鼠(Mus musculus)、牛(Bos taurus)、狐狸(Propithecus coquereli)、骆驼(Camelus ferus)、豹(Pantherophis guttatus)、海龟(Dermochelys coriacea),以分析变异氨基酸的保守性。根据美国医学遗传学与基因组学会(American College of Medical Genetics and Genomics, ACMG)指南,评估变异的致病性。
1.2.4 蛋白模型分析:采用Swis-Pdb Viewer 4.0.1软件构建变异前后的蛋白空间模型,分析变异前后氨基酸相互作用力的变化(PDB,hppt://www.rscb.org/pdb/home/home.do,PDB ID:1AUT)。
2 结果
2.1 基因变异检测结果 全外显子检测提示,先证者PIK3R2基因(NM-005027)存在c.1117G>A杂合变异(见图3A),造成PIK3R2蛋白第373位的甘氨酸替换为精氨酸。先证者父母均未检测到相同变异(见图3B、图3C),提示其为新发(de novo)。该变异在国外有相关报道,但国内尚无报道,本案例为国内首例报道。
2.2 生物信息学分析结果 c.1117G>A变异位于PIK3R2基因的第10号外显子。该变异位于SH2区域(见图4A)。PolyPhen-2软件预测c.1117G>A为可能致病;Mutation Taster、SIFT、CADD、PROVEAN软件预测c.1117G>A为致病。由PIK3R2编码的P85β调节亚基,有4个重要的结构域:一个Src同源3 (Sh3)结构域和一个Rho-Gap同源区域和两个Src同源2(Sh2)区域。c.1117G>A变异导致的p.Gly373Arg变异位于SH2结合域(见图4B)。用Clustal X软件对7种同源物种(哺乳类)的氨基酸序列保守性进行比对,结果表明该位点在8个物种中高度保守(见图5)。 根据ACMG指南,判断该变异为致病性(PS2-Very_Strong+PS4+PM1+PM2+PP3)。
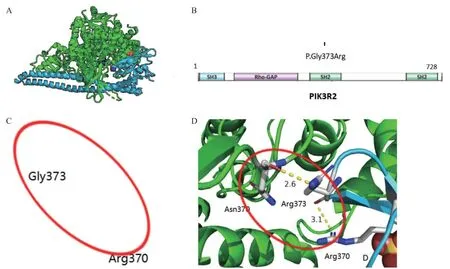
图4 PIK3R2蛋白的结构模型以及野生型和变异蛋白中变异位点的局部结构

图5 PIK3R2基因c.1117G>A变异的进化保守性分析
2.3 蛋白模型分析 用Swiss-Pdb Viewer软件构建野生型与变异型蛋白的结构模型。野生型PIK3R2蛋白Wild 373与Arg370形成一个氢键,维持了该区域的稳定结构,见图4C。该位点发生变异后,Gly373Arg与PIK3CA(绿色)的370Asn之间又形成一个氢键,见图4D,改变了蛋白结构,影响PIK3R2蛋白三位结构的稳定性,从而改变其功能和(或)活性。
3 讨论
MPPH是一种罕见的常染色体显性遗传病,国外已报道近16例由PIK3R2变异引起的MPPH I,但国内目前未见报道;因此,含本例在内目前共17例报道,其中最常见p.Gly373Arg变异15例;罕见变异包括p.Leu401Pro变异1例和p.Asp557His变异1例。MPPH患者临床表现为巨脑畸形,多小脑回以及大脑皮层畸形,部分患者存在癫痫发作、全面发育迟缓以及智力障碍等并发症。见表1。本例先证者因先天性脑积水就诊,表现为特殊面容、巨脑、智力、运动及语言发育相对落后,上述表型与文献报道相符[2-14]。 先证者PIK3R2基因存在c.1117(exon10)G>A杂合变异,通过Mutation Taster等软件预测为致病性。先证者父母表型正常,相应位点未发现变异,符合常染色显性遗传的特点。
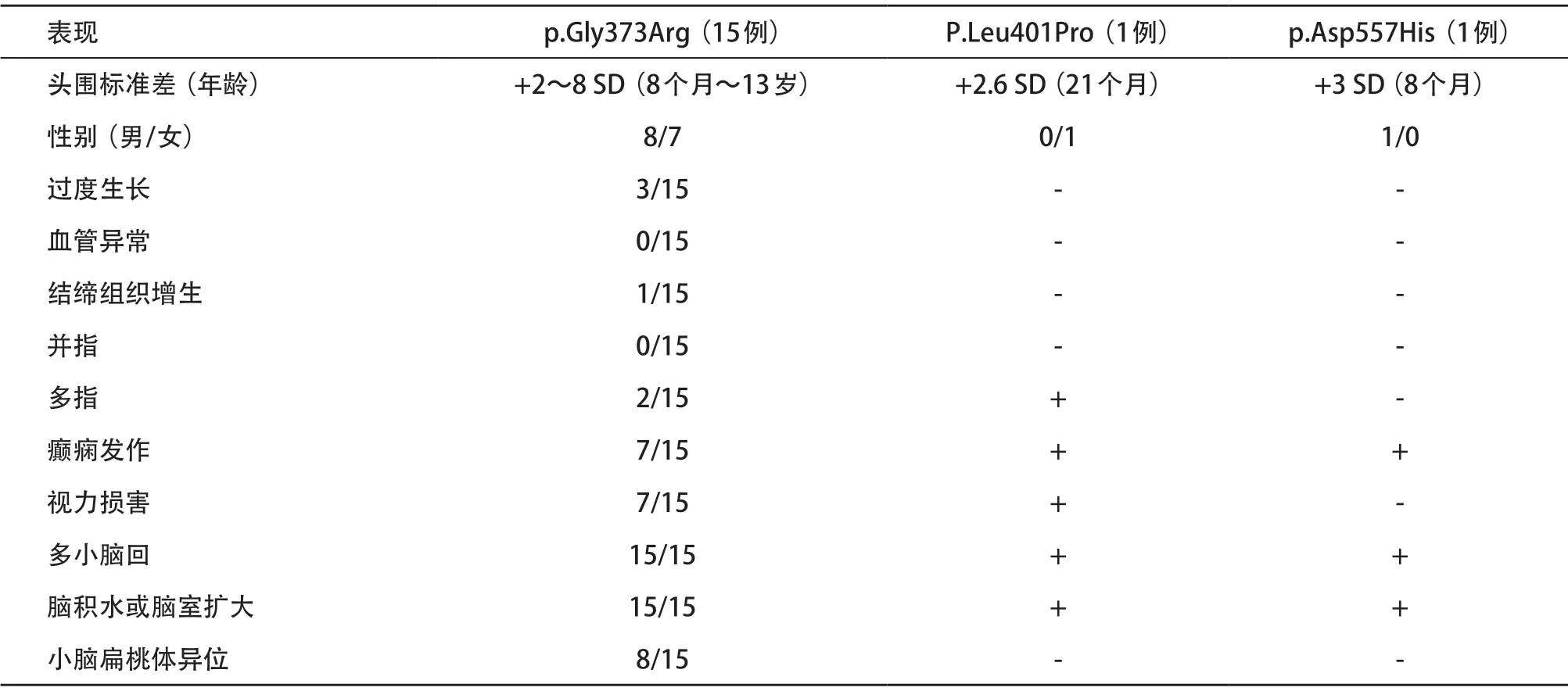
表1 包括本例在内的17例MPPH I型患儿的临床表型
MPPH的临床表型和巨脑-毛细血管畸形-多小脑回综合征(megalencephaly-capillary malformation-polymicrogyria syndrome, MCAP)非常相似。两者是一组共同发育相关的脑发育异常综合征[6,15],前者主要表现为进行性大脑、多小脑回和多指(趾)畸形,后者主要表现为进行性巨脑、多小脑回、毛细血管畸形、并指和结缔组织发育不良。MCAP和MPPH综合征的大部分主要特征可分为5类:①早期过度生长;②血管异常;③远端肢端异常;④大脑皮质发育畸形;⑤结缔组织发育不良。这两种综合征的巨脑畸形都相当显著,头部大小高达 10 SD。虽然这些综合征的表型在很大程度上是重叠的,但可以根据躯体特征加以区分,因为与MCAP相比,MPPH缺乏皮肤血管形成、躯体过度生长、结缔组织发育不良和并指等显著躯体畸形。MCAP可与非对称性脑过度生长(半巨脑)或身体过度生长(节段性过度生长或半肥大)有关,多数MPPH患者的大脑和身体受累呈对称。仅报道的MPPH患者的躯体特征包括:约40%的患者出现轴后多指(趾)畸形,以及轻微的面部畸形特征,如突出的额头和低鼻梁等,这些特征大多继发于脑组织的增加。MCAP与许多出生时就出现的其他临床表型有关,例如皮肤血管畸形,特别是面部和皮肤的毛细血管畸形;并指和多指;结缔组织发育不良和局灶性或节段性身体过度生长;而MPPH除了轴后多指畸形(在报道的个体中约50%),目前报道的案例一致性躯体特征尚不多。由于MPPH的临床表现异质性,目前没有确定的临床指南,诊断相对困难,因此有赖于基因检测技术,如MPPH可能是由PIK3R2、AKT3和CCND2突变引起的;而MCAP与PIK3CA突变有关。本例患儿全外显子检测提示PIK3R2基因c.1117G>A(p.Gly373Arg)错义变异,可排除MCAP综合征,且本案例为国内首例报道。
PIK3R2基因位于19p13.11区,编码由728个氨基酸组成的PIK3R2蛋白。对人、黑猩猩、牛、家属、骆驼等多个哺乳动物物种的PIK3R2蛋白的氨基酸序列进行的比对提示,其第373位的甘氨酸高度保守。国外已有多例携带PIK3R2基因变异的家系报道,其常见的PIK3R2变异位点包括p.Asp557His、p.Gly373Arg和p.Leu401Pro。本例发现的变异位点c.1117G>A(p.Gly373Arg)最为常见。
既往文献报道,磷脂酰肌醇-3-激酶(PI3K)-AKT-mTOR通路(mTOR-通路)[16-17]中的基因变异会导致广泛的大脑和身体发育障碍。MPPH和MCAP的发病均与该通路的过度活化导致过度生长障碍有关,故两者的临床表现巨脑存在显著的重叠。PIK3R2基因位于19号染色体,编码IA类PI3K的P85β调节亚单位[18],是mTOR途径的上游组成部分[19]。PI3K是独特且高度保守的细胞内脂质激酶家族的成员[20], 可磷酸化磷脂酰肌醇和磷酸肌醇的3’-羟基,该反应导致许多错综复杂的细胞内信号通路的激活,包括PI3K-AKT-mTOR通路[21],在细胞的增殖、生长、新陈代谢、迁移和分泌中起着重要作用。P85β调节亚基通过调节催化亚基的稳定性、构象和定位来调节PI3K的活性[22]。P85β调节亚基由一个Src同源3(Sh3)结构域和一个Rho-Gap同源区域和两个Src同源2(Sh2)区组成,这两个区域通过一个称为SH2间区(i-sh2)的螺旋线圈区域相连,以介导P85β调节亚基与催化亚基的结合[12,14,19]。其中Sh2结构域是一个约100个氨基酸残基的蛋白质结构,作为细胞内信号级联的调节模块,通过与含有磷酸酪氨酸的靶肽在序列特异性中的高亲和力相互作用,识别磷酸化酪氨酸的3~6个残基C末端,并严格依赖于磷酸化方式。有研究[11]成功建立了人类最常见的PIK3R2变异p.G373R(小鼠p.G367R)小鼠模型。这些小鼠中,显示PI3K-AKT-mTOR通路被过度激活和脑过度生长,小鼠出现的巨脑和脑电变化都与人类表型重叠。证实了PI3K复合体的变异与广泛的脑和器官过度生长综合征有关,促进细胞生长可能是通过蛋白质S6磷酸化增加介导的蛋白质合成增加所致。PIK3R2变异诱导的mTOR-通路激活,导致大脑细胞增大,是临床表型相关巨脑最可能的机制。本案例先证者的p.Gly373Arg变异位于SH2结构域,导致带中性电荷的甘氨酸被带正电荷的精氨酸取代后,蛋白质的局部理化性质可能会发生变化。另外变异后形成新的氢键,结构的改变可能会进一步导致蛋白质功能的变化。同时该患儿存在语言认知发育及运动相对落后,但无癫痫发作,无轴后多指等畸形,与既往文献报道表型存在异质性,考虑如下:①不同的基因变异导致不同的表型,如本例PIK3R2致病变异位于SH2-1结构域,而既往文献报道的部分病例位于SH3结构域附近。②基因的功能受到多种因素的影响,包括翻译后修饰、基因-基因相互作用、环境等。③另外,目前该基因变异以国外报道为主,推测可能存在不同种族基因表型共分离。因此,同一基因的变异可能会导致不同个体的不同结果。综上,PIK3R2基因第10外显子c.1117G>A(p.Gly373Arg)杂合变异可能是本例患者罹患MPPH的原因,其确切的致病机制仍需进一步的基因表达和功能实验加以验证。
猜你喜欢
临床输血与检验(2022年3期)2022-06-22 02:52:50
临床小儿外科杂志(2021年2期)2021-03-04 05:20:12
郑州大学学报(医学版)(2019年3期)2019-06-03 06:19:32
传染病信息(2019年2期)2019-05-17 13:16:04
现代园艺(2017年21期)2018-01-03 06:41:32
中国康复理论与实践(2015年10期)2015-12-24 05:42:44
医学研究杂志(2015年6期)2015-07-01 17:40:30
医学研究杂志(2015年5期)2015-06-10 06:43:26
当代畜禽养殖业(2015年4期)2015-03-23 07:15:45
重庆医学(2015年12期)2015-03-05 05:52:54
