
院内制剂夏膝颗粒中迷迭香酸含量的HPLC测定
2022-12-10 08:39成光宇位鸿何蕊
长春中医药大学学报 2022年12期
陈 珍,成光宇,位鸿,何蕊,陈 颖*
(1长春中医药大学,长春 130117;2长春中医药大学附属医院,长春 130021)
夏膝颗粒处方为首届国家名中医黄永生教授的临床经验方,用于治疗眩晕之痰瘀互结证型的高血压,于临床上取得良好的疗效,将其开发成院内制剂—夏膝颗粒,本制剂由夏枯草、牛膝、茺蔚子、决明子等八味药组成,夏枯草为本方中的君药,现代药理学实验证明夏枯草具有良好的降压作用[1],其中迷迭香酸是夏枯草中的有效成分[2-3]。本试验以夏枯草中迷迭香酸对照品作为对照,研究并建立了HPLC迷迭香酸的含量测定方法,可以作为夏膝颗粒质量控制的依据。
1 材料与方法
1.1 仪器与试药
1.1.1 仪器 高效液相色谱仪(日本岛津LC-2030C);紫外检测器(LC solution色谱数据工作站);色谱柱(岛津Inertsil,规格:4.6 mm×250 mm,5 μm);超声波清洗机(昆山禾创超声仪器有限公司); CP225D型电子天平(德国sartorius公司)。1.1.2药品与试剂 迷迭香酸对照品(批号:111871-202007,规格:每支20 mg,采购于中国食品药品检定研究院);甲醇为色谱纯,水为超纯水;其它试剂均为分析纯。供试样品:夏膝颗粒(批号:20220101、20220102、20220103)。
1.2 色谱条件与系统适应性
十八烷基硅烷键合硅胶为填充剂;流动相:甲醇-0.1%三氟乙酸(40:60);检测波长330 nm;流速1.0 mL·min-1。进样量5 µL;迷迭香酸的理论塔板数不低于6 000[4];柱温为25 ℃。
1.3 对照品溶液的制备
精密称取对照品迷迭香酸10.06 mg,置10 mL量瓶中,加稀乙醇至刻度,摇匀,得浓度为1.006 mg·mL-1的溶液;再精密吸取上述溶液1 mL,置10 mL量瓶中,加甲醇至刻度,摇匀,得浓度为0.1006 mg·mL-1的对照品溶液[4]。
1.4 供试品溶液的制备
取本品粉末(过四号筛)约4 g,精密称定,置具塞锥形瓶中,精密加入稀乙醇50 mL,超声处理(功率240 W,频率50 kHz) 30 min,放冷,再称定重量,用稀乙醇补足减失的重量,摇匀,滤过,取续滤液,即得[4]。
2 方法学验证
2.1 专属性考察
精密吸取对照品溶液、供试品溶液、阴性对照溶液各5 mL,注入液相色谱仪,色谱图见图1、2、3。
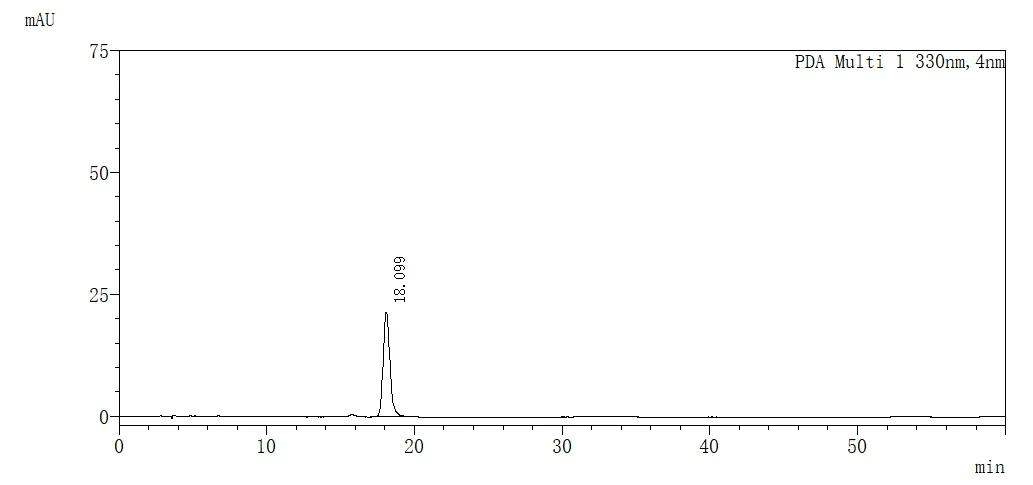
图1 迷迭香酸对照品高效液相色谱图

图2 供试品溶液高效液相色谱图
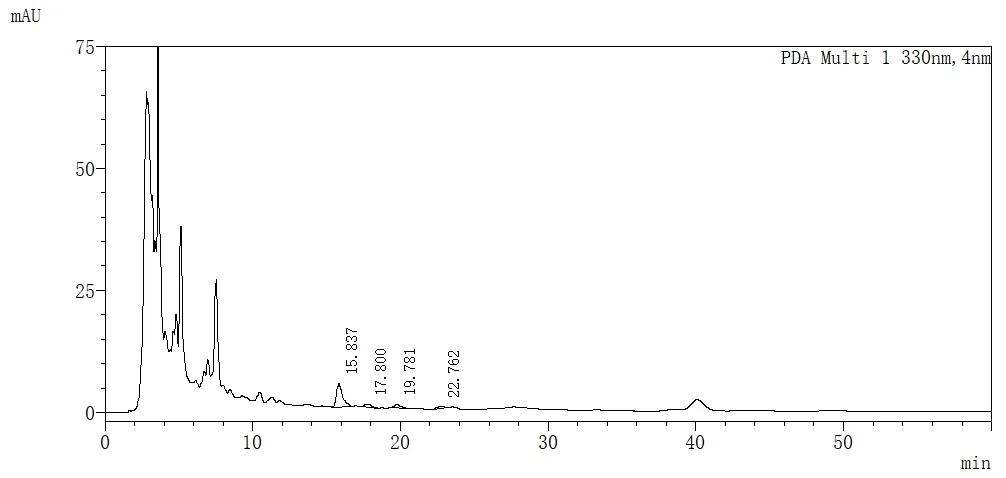
图3 阴性对照溶液高效液相色谱图
可见在此条件下,迷迭香酸峰与其它峰分离较好,阴性对照无干扰。
2.2 线性关系考察
分别精密吸取迷迭香酸对照品溶液1 mL,2 mL,5 mL,10 mL,15 mL,20 mL,注入液相色谱仪,测定,以进样量(mg)为横坐标,峰面积积分值为纵坐标,绘制标准曲线。
回归方程:Y = 2 799 488.530 8 X-3 981.079 0,r = 1.000 0 ,迷迭香酸线性范围为0.050 3 mg~1.006 0 mg。结果见图4。

图4 迷迭香酸标准曲线
2.3 耐用性考察
2.3.1 流速的考察 选择0.8 mL·min-1、1.0 mL·min-1、1.2 mL·min-1的流速进行试验,含量测定结果:RSD为0.51%,说明流速对测定结果影响不大。结果见表1。
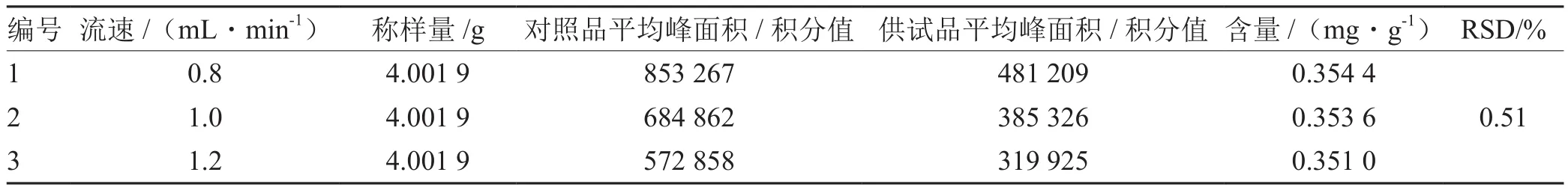
表1 流速考察结果
2.3.2 柱温考察选择25℃,30℃,35℃,依法测定,考察柱温对分离效果的影响,测定结果:迷迭香酸的RSD为5.58%(见表2)。其中25℃,30℃条件下迷迭香酸RSD为1.60%,但25℃条件下供试品的峰是融合峰,基线未完全分离;30℃,35℃条件下迷迭香酸RSD为7.40%,说明柱温对含量测定结果影响较大,柱温要控制在30℃。

表2 不同柱温考察结果(25℃,30℃,35℃)
2.3.3 不同色谱柱的考察选取不同品牌的色谱柱考察其对试验结果的影响,RSD为0.19%,说明不同色谱柱对其测定结果影响不大。结果见表3。

表3 不同色谱柱考察结果
综上所述,通过对流速、柱温、不同流动相比例和不同品牌色谱柱的考察试验,发现流速和不同品牌色谱柱的变动测定结果影响较小,柱温和不同流动相比例对测定结果影响较大,在试验中需要控制。
2.4 稳定性考察
精密吸取对照品溶液和同一供试品溶液各5 mL,分别在0 h,4 h,8 h,12 h,16 h,24 h依法测定,以迷迭香酸峰面积积分值为指标。结果见表4。
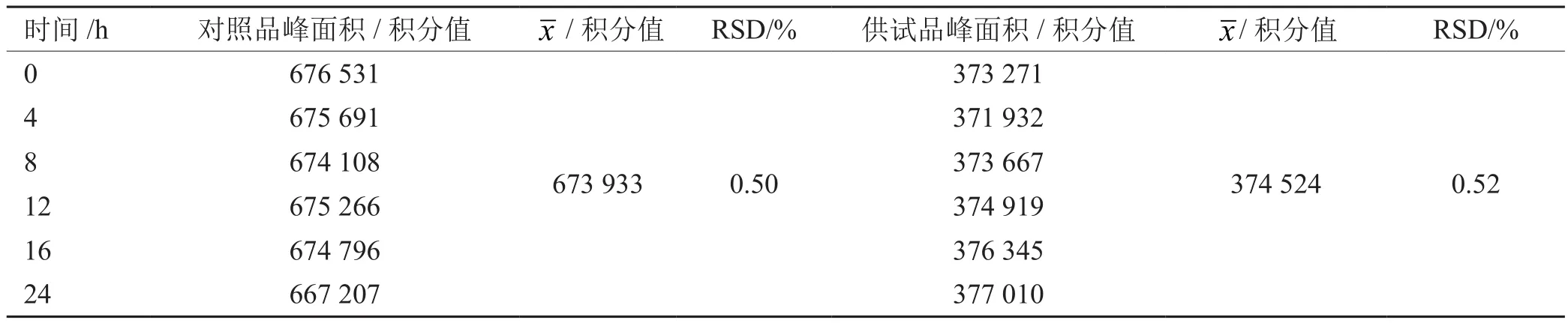
表4 稳定性考察结果
结果:对照品溶液的RSD为0.50%,供试品溶液的RSD为0.52%,24 h内稳定性良好。
2.5 精密度考察
精密吸取对照品溶液和同一供试品溶液各5 mL,注入液相色谱仪,连续进样6次。结果见表5。

表5 精密度考察结果
精密度考察结果显示:对照品溶液的RSD为0.21%,供试品溶液的RSD为0.10%,方法、仪器的精密度良好。
2.6 重现性考察
精密称取同一批样品6份,照1.4供试品溶液项下制备方法制备,依法独立测定。结果见表6。
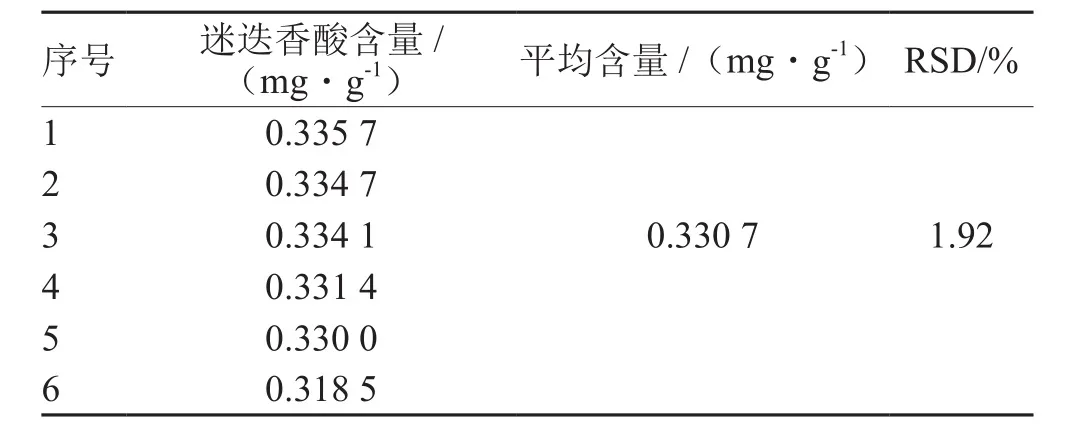
表6 重现性考察结果
考察结果:RSD为1.92%,平均含量为0.330 7 mg·g-1,此方法的重现性良好。
2.7 准确度试验
供试品的制备:取重复性试验同批样品2 g,研细,精密称定,置于具塞锥形瓶中,加0.704 2 mg·mL-1的迷迭香酸溶液1 mL,加入稀乙醇49 mL,超声处理30 min,放冷,称定其重量,用稀乙醇补足减失的重量,摇匀,滤过,取续滤液,即得。共制成6份样品,依法测定,计算回收率。结果见表7。

表7 回收率试验结果
测定结果:RSD为1.47%,平均回收率为97.72%,本法准确性较好,方法可行。
2.8 含量测定
取三批中试样品,依法制备供试品溶液,依法测定。结果见表8。
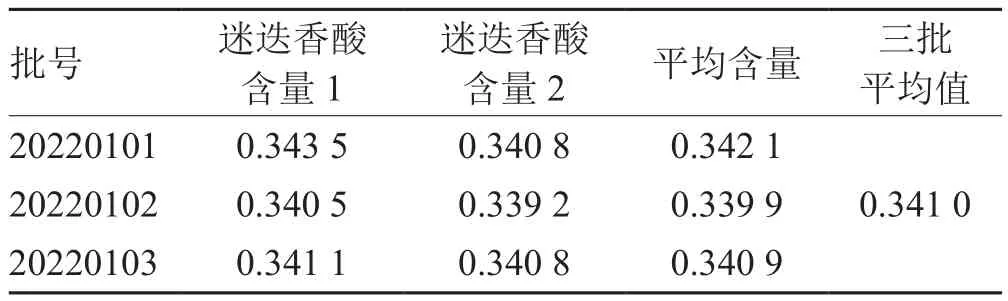
表8 三批中试含量测定结果 mg·g-1
故暂定:本品每袋颗粒含夏枯草以迷迭香酸(C23H28O11)计,不得少于2.40 mg。
本品三批中试样品含量测定的平均结果为0.341 0 mg·g-1,本品规格为每袋装10 g,含量限度按测定结果平均值的80%计算:制剂中迷迭香酸含量×每袋装量×80% = 0.341 0 mg·g-1×10 g/袋×80% = 2.727 9 mg/袋。但投料时,每批夏枯草药材的含量有高有低,也可以按中国药典规定的夏枯草药材的含量和提取转移率计算,根据正交试验的验证性试验结果,迷迭香酸的平均转移率70.53%(按验证的转移率计算),按《中国药典》夏枯草项下“含迷迭香酸(C23H28O11)不得少于0.30%”的规定[4],标准处方中,夏枯草220.0 g,制成1 000 g,本品规格为每袋装10 g,含量限度按测定结果平均值的80%计算,本品每袋颗粒含夏枯草以迷迭香酸的计,计算公式:夏枯草药材量×夏枯草药典含量×平均转移率×每袋装量×80%÷标准处方量= 220 g×0.20%×70.53%×10 g/袋×80%÷1 000 g = 2.48 mg/袋
3 结论
药典以甲醇-0.1%三氟乙酸(42:58)为流动相,在此条件下,供试品色谱中迷迭香酸色谱峰与相邻f峰分离欠佳,将流动相比例调整为甲醇-0.1%三氟乙酸(40:60),迷迭香酸色谱峰分离良好。
本试验以夏枯草中迷迭香酸对照品为对照,进行了方法学考察并建立了含量测定的方法,暂定:本品每袋颗粒,含夏枯草以迷迭香酸(C23H28O11)计,不得少于2.40 mg。
猜你喜欢
人人健康(2021年14期)2021-08-06
粮食储藏(2021年6期)2021-03-29
家庭医学(2020年8期)2020-09-02
时代邮刊(2019年22期)2019-12-17
时代邮刊·下半月(2019年11期)2019-09-22
——CIPAC分析手册 O卷的分析方法
农药科学与管理(2018年2期)2018-08-07
家庭医学(2017年9期)2017-10-24
家庭百事通·健康一点通(2017年7期)2017-07-04
中国民族医药杂志(2016年4期)2016-05-09
分析化学(2015年6期)2015-06-18
