
发作性运动障碍相关基因突变与癫痫相关性的研究进展
2022-12-09 12:35:04黄昕祺亓蕾任连坤
癫痫与神经电生理学杂志 2022年5期
黄昕祺,亓蕾,任连坤
发作性运动障碍(paroxysmal movement disorders,PMD)多见于儿童,是一类以发作性不自主运动为主要特征的遗传病[1]。该病通常分为两种类型:发作性异常运动 (paroxysmal dyskinesia,PxD) 和发作性共济失调 (episodic ataxia,EA)[2]。PxD根据诱发因素和临床特点又可进一步分为发作性运动诱发性运动障碍 (paroxysmal kinesigenic,PKD)、发作性非运动诱发性运动障碍(paroxysmal non-kinesigenic,PNKD)及发作性过度运动诱发性的运动障碍(paroxysmal exercise-induced dyskinesia,PED)[3],详见表1。发作性睡眠诱发运动障碍 (paroxysmal hypnogenic dyskinesia,PHD) 曾被定义为PxD的第4种类型,后被证明是一种夜间发作的常染色体显性遗传的额叶癫痫[4]。
PMD在被报道后的很长一段时间里都被认为是癫痫的一种发作形式。随着神经遗传学的快速发展,越来越多的研究发现癫痫和PMD在基因型、表型及治疗等方面存在着广泛的交叉。
1 PMD概述
1892年,Kure首次报道了1例由运动诱发出异常运动的日本青年,该患者被诊断为“Thomsen′s disease”。Gowers在1901年也描述了类似的症状,但将其归因为癫痫的一种特殊发作表现。1940年,Mount 和Reback报道了1例不自主肢体和躯干扭动的患者并命名为发作性舞蹈徐动症,之后被证明是PNKD。1946年,Parker发表了首个发作性共济失调病例。Kertesz和Weber于1967年将此类不自主运动病史的家族称为发作性舞蹈徐动症和家族性发作性肌张力障碍。1977年,Lance描述并命名了第1例PED。1995年,Demirkiran等汇总多方术语提出将PMD分为PKD、PNKD及PED 3种亚型,该分类标准广为流传并沿用至今。1994年前后,EA1和EA2陆续被证明分别与KCNA1基因和CACNA1A基因的突变相关。2004年,MR-1基因(后更名为PNKD基因)被证明其突变与PNKD的发生相关。2008年,研究者们在PED患者中观察到SLC2A1基因的突变。2011年,PRRT2突变型被报道为PKD的致病基因。除此之外,还有许多其他的基因型被陆续证明与PMD和癫痫共相关。
2 PMD与癫痫
尽管PMD与癫痫在临床表现及病理生理机制等方面都有相似之处,但它们仍是两种独立的疾病。PMD的影像学检查和脑电图多正常,必要时可借助基因学检查明确诊断,但同时需注意存在与癫痫共病的可能[5-6]。
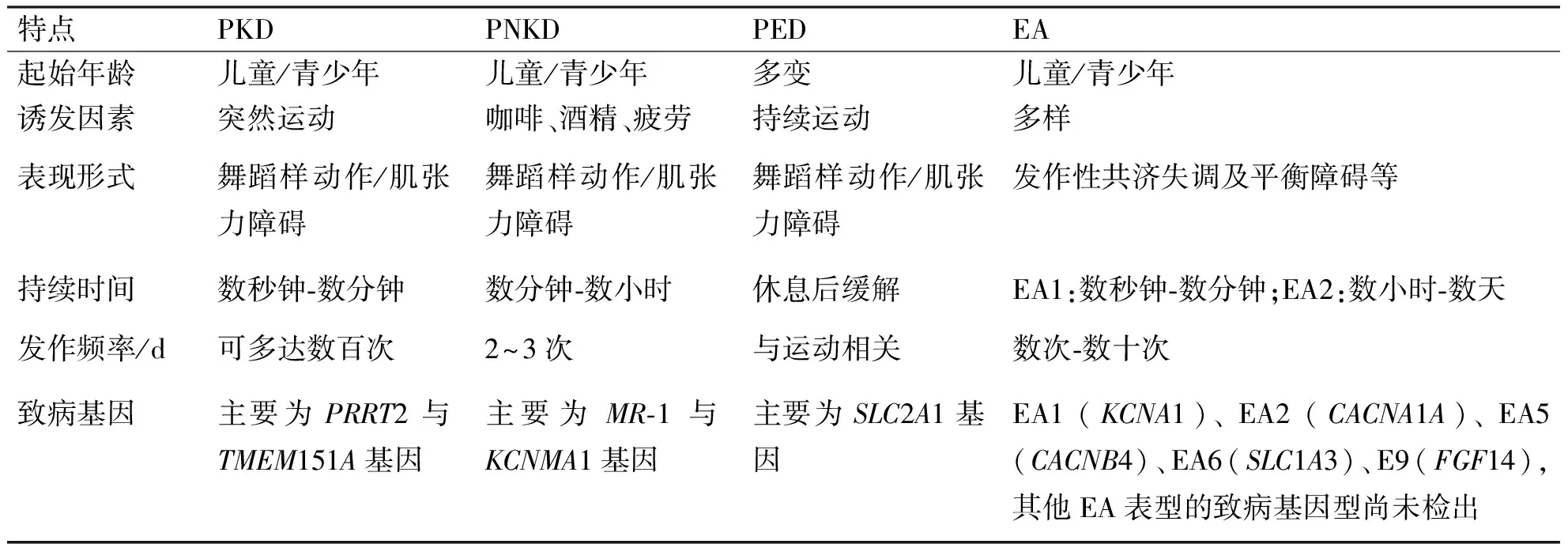
表1 各型PMD的临床特点
2.1 PKD和癫痫
PKD又称发作性运动诱发性舞蹈手足徐动症,是PMD中发病率最高的一种亚型,多由突然的运动、惊吓或过度换气诱发,发作持续数秒钟后自行缓解,一天可发作上百次。患者出现姿势性肌张力障碍或舞蹈样动作,发作时意识清楚,神经系统检查与脑电图多无异常且缺乏发作后状态。最新中国指南将PKD分为原发性和继发性两类,并将基因检测定为Ⅰ级推荐诊断方法。原发性PKD主要由PRRT2基因和TMEM151A基因突变致病,PRRT2突变型和癫痫关联密切。继发性PKD多由多发性硬化、头外伤及假性甲状旁腺功能减退等疾病引发[7]。同时,PRRT2突变也是PKD和良性家族性婴儿癫痫(benign familial infantile seizure,BFIS)最常见的病因,由PRRT2基因编码的PRRT2是富含脯氨酸的一种跨膜蛋白,突变后可影响神经递质的释放[8]。BIFS患者常常合并PKD,两者发作短而频繁,且多发生于热性惊厥的背景下,对小剂量卡马西平及苯妥英钠等抗癫痫药物(antiepileptic drugs,AED)反应良好。临床上将该类共患病合称为婴儿惊厥性舞蹈病综合征 (infantile convulsions and choreoathetosis,ICCA)[9]。SCN8A突变也可导致PKD的发生,同时也被证明与发育性癫痫性脑病相关[10]。而PKD的其他基因型如DEPDC5、KCNA1、KCNMA1、SLC2A1及PNKD基因,其突变最初都在具有可变病灶的局灶性家族性癫痫的背景下描述[11]。CHRNA4基因与PKD、癫痫和发育迟缓共相关[12]。
2.2 PNKD和癫痫
PNKD多于儿童期起病,由咖啡、疲劳及精神刺激等非运动因素诱发,发作时间较PKD长,常持续数十分钟甚至几小时,发作频率较低,每天仅发作1~3次,并可有数月的间隔期。PNKD发作前可有感觉异常先兆,发作时语言功能受累但意识清楚[13]。原发性PNKD的发生与MR-1基因突变强关联,该突变导致胞内的mRNA失去c端的PNKD-1,其抑制胞吐的功能减弱,囊泡释放增加,导致了PNKD的发生,目前暂未发现该突变与癫痫的关系[14]。有研究报道了1例典型的携带MR-1基因突变的PNKD患者,氯硝西泮在一定程度上减缓了疾病的进展[5]。PNKD的其他致病基因还包括KCNMA1、ATP1A3及GNAO1基因等。KCNMA1基因编码Ca2+激活的无K+通道亚单位,与癫痫和发育延迟相关[15]。编码钠钾泵的ATP1A3基因已被证明其突变是儿童交替性偏瘫 (alternating hemiplegia of childhood,AHC) 的主要病因,而AHC又以进行性认知和运动障碍以及频发癫痫为主要临床表现[16]。GNAO1基因编码G 蛋白α亚基多肽链O,突变后可影响cAMP途径,使患者出现癫痫性脑病和发育迟缓[17]。
2.3 PED和癫痫
PED是PMD中发病率最低的一种类型,目前报道PED的研究数量很少。PED通常发生在持续运动后,患者出现发作性肌张力障碍及手足徐动等不自主运动,运动肢体(下肢)最常受累。发作持续5~30 min,停止运动和生酮饮食可缓解症状。PED由编码葡萄糖转运蛋白1 (Glut-1)的SLC2A1基因 (GLUT1基因) 突变导致,突变后的Glut-1蛋白对葡萄糖的转运能力显著下降,患者出现运动障碍、癫痫、神经发育迟缓及低血糖等一系列疾病[18]。功能成像研究发现与PED相关的皮质纹状体通路和与癫痫相关的额叶皮质中都存在葡萄糖代谢改变,这也解释了SLC2A1基因突变可以同时产生PED和癫痫的原因[19]。除SLC2A1基因外,ECHS1、ATP1A3、TBC1D24及GCH1基因都曾被报道与癫痫和PED共相关[20-22]。这些研究均证明了SLC2A1突变绝不是PED的唯一病因。
2.4 EA和癫痫
EA是一种罕见的常染色体显性遗传的离子通道病,可由多种因素诱发。EA以发作性共济失调为主要表现,常合并震颤、眩晕、构音障碍及肌张力障碍等其他症状[23]。目前的研究把EA分为9个表型 (EA1-EA9),除EA1和EA2外,其他EA表型的临床表现都十分相似,主要依靠基因检查鉴别。EA1由KCNA1基因的杂合突变引发,通常不伴有眩晕、眼球震颤和头痛,间歇性肌痉挛是该病的特征之一。约10%的患者在婴儿期或儿童期同时出现癫痫,表现为部分性或全身性发作,仅少数合并有癫痫性脑病。CACNA1A为EA2的致病基因,眩晕、偏头痛及眼震是EA2的常见症状。部份EA2患者合并癫痫,表现为失神发作、强直阵挛发作及癫痫性脑病等多种组合。EA3、EA4的眩晕和共济失调症状突出,虽有文献记载,但尚未检测出其致病基因。CACNB4基因编码的电压门控Ca2+通道突变同时增加了发生EA5和癫痫的易感性。EA6首次在1例患有发作性共济失调、癫痫及交替性偏瘫的男孩中记录到,病因是SLC1A3基因突变影响了谷氨酸能突触中的神经递质浓度调节。Conroy在2017年首次报道了EA8,抗癫痫药物氯硝西泮可改善患者的症状[24]。近年来有研究发现了由编码成纤维细胞生长因子的FGF14基因突变致病的EA9[25]。
3 PMD与癫痫的治疗

4 结语和展望
随着现代基因学日新月异的发展,按照现象学和病因学来区分疾病表型的传统观念也受到越来越多的挑战。针对PMD和癫痫的高度相似性,仅依靠临床表现和常规检查手段来鉴别诊断存在一定的困难,遗传学检查或可成为更加有效的诊断标准和方式。早期基因确诊可以最大限度地争取治疗时间,节约诊断成本,避免肌肉活检及脑组织活检等侵入性检查。不仅如此,精确的基因检测结果还可以针对致病的基因突变采取相对最有效的治疗方式,更可以通过产前检查为下一代的健康做好保障,利用先进的辅助生殖技术来规避致病基因。相信在不远的将来,基因学的发展会让更多表型的致病基因浮出水面,在PMD和癫痫的发病机制、诊断及治疗上取得更大的突破。
猜你喜欢
英语世界(2023年6期)2023-06-30 06:29:10
中国神经精神疾病杂志(2022年3期)2022-07-14 02:23:42
世界科学技术-中医药现代化(2021年8期)2021-12-21 07:04:52
中国生殖健康(2020年2期)2021-01-18 02:51:26
小学生导刊(2018年13期)2018-06-29 03:49:00
现代园艺(2017年21期)2018-01-03 06:41:32
中国医药科学(2015年24期)2016-03-07 15:32:46
中国康复理论与实践(2015年10期)2015-12-24 05:42:44
医学研究杂志(2015年5期)2015-06-10 06:43:26
现代检验医学杂志(2015年5期)2015-02-06 01:42:20
