
磁性Co/TiB2室温高效催化氨硼烷产氢及串联降解有机污染物
2022-12-06 06:29宋俊杰贾洪柏丁宏云凡王金智康杜向博文李仁宏
无机化学学报 2022年11期
宋俊杰 韦 童 许 超 贾洪柏 刘 军 丁宏云 何 凡王 敏 金智康 杜向博文 王 刚 李仁宏*,
(1浙江理工大学材料科学与工程学院,杭州 310018)
(2安吉国千环境科技有限公司,湖州 313300)
0 引言
近年来,化石燃料的过度使用加剧了日益严重的能源危机和环境问题,清洁新能源的开发成为当下最重要的研究课题之一[1-3]。氢能由于其高燃烧热值以及水作为唯一产物等优点,被认为是21世纪最理想的清洁能源[4-5]。但氢气极低的临界温度和密度导致氢能的高效储运仍然是其利用的瓶颈。为了实现以燃料电池为主的“氢经济”和改善环境问题,开发一种从可再生、低污染资源中高效、环保地制取氢气技术具有重要意义[6-9]。
在众多新型储氢材料中,作为硼氮化合物的氨硼烷(NH3BH3)具有较高氢含量(19.6%)、低分子量(30.87)、无毒、在空气和水溶液中的稳定性高等优点。传统的化石燃料制得的为灰氢(生产过程有CO2等的排放),并且随着天然气等化石燃料的价格波动而产生更多的CO2减排成本[10]。而NH3BH3分解气相产物只有氢气,无需进行副产物如CO2的捕获提纯,从而在节约制氢成本同时实现碳的零排放,符合绿氢理念,具有一定经济性,已成为一种很有前途的储氢材料[11-13]。NH3BH3脱氢可以通过溶液形式的水解反应或者固态形式的热解反应进行,其中水解反应在加入合适的催化剂后可在室温下可控地产出氢气,1 mol NH3BH3可以水解产出3 mol氢气,即。基于此,其催化水解制氢受到广泛关注[14-15]。
另一方面,串联催化技术在高效催化领域中已经取得重要进展。所谓串联催化是将多种试剂和/或催化剂在一个反应容器中进行一系列的阶段性催化反应,为构建新型催化体系提供了新途径[16],如Luo等[17]将石墨烯量子点(GQDs)与掺杂钒的二氧化钛结合制备的GQDs/V-TiO2催化剂,可以光催化降解亚甲基蓝染料并将产生的CO2进一步光催化转化为甲醇、乙醇和甲烷等有用的有机化合物。我们此前研究发现,催化产氢体系可进一步串联有机污染废水的降解处理,并且无需添加光、热能等外部能量或H2O2等助剂,创新性地在新能源和污水处理两大领域之间搭建了一座桥梁[18-20]。废水中除含有无机污染物外,更含有大量的有机污染物,这些有机污染物会在自然环境中积聚,最终危害人类的健康[21]。有机污染物中硝基苯酚和印染染料的直接排出会对水体产生极度危害,被认为是优先处理污染物。硝基苯酚在农业和工业中的大规模应用导致在水体中过量存在,人或动物不小心饮用将会对肝脏、肾脏、中枢神经系统以及血液等产生不良影响[22-23]。另外,根据文献调研,全球约有10 000种染料用于印染行业,每年工业生产的染料量约为70万吨,其中10%~15%的染料被直接排放到水体中造成污染[24-25]。此外,在各种染料中,偶氮染料被发现是毒性更强的染料,其官能团偶氮基(—N=N—)具有致癌作用,未经处理的偶氮染料废水直接排放到环境中极有可能严重影响接触者的健康[26]。
考虑到先前研究使用的Pd、Ag等贵金属催化剂具有高成本和不易回收性,便于回收利用的磁性非贵金属纳米催化剂成为了更好的解决方案。基于此,本工作采用简单的沉淀-沉积法制备Co/TiB2磁性纳米催化剂,用于室温下催化NH3BH3水解产氢。高温还原气氛处理后Co纳米粒子被TiB2载体包覆,具有典型的金属-载体强相互作用(SMSI),得益于彼此间强的电子传递,Co/TiB2催化产氢速率最高可达565.8molH2·molcat-1·h-1。另外,我们发现该催化产氢体系在不添加外部能量或者额外助剂下即可高效串联降解有机污染物,如对硝基苯酚(4-NP)和典型的偶氮染料如酸性橙7(AO7)、酸性红1(AR1)和甲基橙(MO)。反应结束后通过引入外加磁场即可快速回收催化剂,避免其对水体造成二次污染,且制备方法和反应过程相对绿色环保,为开发催化制氢技术与有机污染废水处理提供了绿色、低能耗的新策略。
1 实验部分
1.1 试剂
所用到的试剂有六水合硝酸钴、纳米二氧化钛、无定形硼粉、氯化钾、氯化钠、无水乙醇、氢氧化钠、氨硼烷(NH3BH3)、4-NP、对氨基苯酚(4-AP)、AO7、AR1、MO。所有试剂均为分析纯,实验过程中无需进一步提纯,溶剂为去离子水(自制)。
1.2 催化剂制备
TiB2的制备:将物质的量之比为3∶10的TiO2与无定形硼粉充分研磨30 min得粉末A,按物质的量之比1∶1称取NaCl与KCl混合粉末B作为熔融盐,随后按照质量比1∶10将粉末A与粉末B充分混合并研磨30 min。将所得混合物放入刚玉坩埚中并置于管式炉中心,在N2气氛下以10℃·min-1的升温速率升温至900℃并在此温度下保温1 h后随炉冷却至室温,将产物放入80℃热水中并通过超声处理清洗除去熔融盐以及B2O3等杂质,随后用无水乙醇离心洗涤,放入60℃烘箱干燥备用。
Co/TiB2磁性纳米催化剂的制备:采用简单的沉积-沉淀法制备了催化剂前驱体,取一定量的六水合硝酸钴与TiB2分散到100 mL去离子水中搅拌30 min,之后在搅拌过程中逐滴加入0.125 mol·L-1的NaOH溶液直至完全形成沉淀,收集后用无水乙醇离心洗涤,放入60℃烘箱干燥过夜,此时得到Co(OH)2/TiB2前驱体。取一定量的前驱体置于石英舟中并置于管式炉中心,在氢氩混合气(φH2=5%)还原气氛下以10℃·min-1的升温速率升温至600℃并保温2 h,然后随炉冷却至室温,获得Co/TiB2催化剂。通过改变前驱体溶液中六水合硝酸钴的用量,以制备不同Co负载量(x%,质量分数)的TiB2催化剂x%Co/TiB2。
1.3 仪器及其工作条件
X射线衍射(XRD)使用的是Burker D8型衍射仪,辐射源是Cu Kα,波长为0.154 nm,工作电压35 kV,工作电流25 mA,扫描范围10°~80°,扫描速度3(°)·min-1。使用蔡司 Sigma 300 型扫描电子显微镜(SEM)观察样品形貌,加速电压为3 kV,能谱(EDS)加速电压为15 kV。透射电子显微镜(TEM)照片采用FEI Tecnai G2 F20型透射电子显微镜获得,最大加速电压200 kV。X射线光电子能谱(XPS)在Thermo Scientific K-Alpha光谱仪上测得,激发源是单色铝靶(Al Kα),hν=1 486.6 eV,结合能均以 C1s结合能(284.80 eV)为能量标准进行电荷校正。磁滞回线使用Quantum Design PPMS-9型振动样品磁强计(VSM)测定,磁场分辨率0.02 mT,磁场强度范围-3~3 T。由产氢性能结果得知,Co的负载量为15%时催化剂性能最佳,因此催化剂的表征以及有机污染物降解测试均使用15%Co/TiB2催化剂(以下简称Co/TiB2)。
使用Bruker EPR A-300电子顺磁共振(EPR)光谱仪在室温条件下检测有机污染物降解过程中产生的自由基。以5,5-二甲基-1-吡咯啉-N-氧化物(DMPO)作为自由基捕获剂,参数设置如下:中心场为3 507 G,扫描宽度为100 G,微波频率为9.86 GHz,信号调制频率为100 kHz,功率为20 mW,转换时间为20 ms。
使用日立UV-3900型紫外可见分光光度计(UVVis)检测降解反应中的有机污染物吸收光谱的变化,采样间隔为0.5 nm,高速扫描。
1.4 催化性能检测
1.4.1 催化产氢性能检测
催化NH3BH3溶液产氢反应在55 mL的石英试管中进行,搅拌速度为(400±10)r·min-1。在试管中添加5 mg的 x%Co/TiB2,加入 5 mL NH3BH3溶液,用橡胶塞塞住试管口,用封口膜密封确保气密性后将其置入搅拌器上的25℃水浴反应30 min,每隔5 min用微量进样针从试管中抽取400µL气体注入GC-TCD气相色谱仪中,检测体系中H2、O2等气体含量。NH3BH3催化产氢的速率计算公式为vH2=nH2/(nCot),其中nH2和nCo分别表示在t时产生的H2和催化剂中Co的物质的量。
1.4.2 催化降解性能检测
1)课前,教师利用智慧课堂手机客户端设计学情分析问卷,帮助学生确立学习目标、制订学习计划,提升学生的自主学习能力。学生通过课前导学案和单词学习软件,按照教师布置的任务开始本单元学习。教师将设计好的以本单元热点教学内容为主题的投票问卷上传到智慧课堂手机学习平台,与学生形成互动,引导学生形成正确的文化意识和思维。
将 40 mL NH3BH3(0.01 mol·L-1)与 4-NP(0.1 mmol·L-1)混合溶液加入容积为50 mL的玻璃瓶中,称取5 mg Co/TiB2加入玻璃瓶中,在室温下进行搅拌。加入催化剂后立刻用注射器抽取2 mL混合液,通过磁铁将催化剂磁分离后将清液加入比色皿中,用UV-Vis测其吸收光谱(对应反应时间0 min),之后每隔一定时间抽取2 mL混合液,经磁分离出催化剂后进行UV-Vis检测,直至反应液变为无色或吸光度达到预设值。偶氮染料的降解性能检测时,将40 mL NH3BH3(0.01 mol·L-1)与偶氮染料(AO7、AR1 和MO,0.2 mmol·L-1)的混合溶液按照上述相同的步骤进行。
根据Lambert-Beer定律计算溶液吸光度与浓度的关系:A0/A=c0/c,其中c0为有机污染物溶液的初始浓度值,c为t时间有机污染物溶液的浓度值,A0为有机污染物溶液初始浓度的最大吸收波长(λmax)处所对应的吸光度,A为t时间λmax处所对应的吸光度。通过不同时间溶液浓度变化来计算有机污染物的降解率R,其计算公式为R=[(c0-c)/c0]×100%。反应速率方程:ln(c0/c)=kt,其中k为反应速率常数。
AO7染料降解循环实验步骤如下:在相同的条件下进行循环降解实验,第1次实验结束后通过磁铁将催化剂磁分离回收后加入到相同量的NH3BH3与AO7染料混合溶液中继续进行第2次实验,反应时间与第1次相同,如此进行多次实验。
2 结果与讨论
2.1 催化剂表征
图1a~1d是磁性纳米催化剂Co/TiB2的SEM图以及EDS元素分布图,可以看出Co纳米粒子均匀地分布在TiB2载体上,无明显的团聚现象。图1e是Co/TiB2的TEM图,进一步解析出晶格间距为0.204和0.263 nm,分别对应于Co的(111)晶面和TiB2的(100)晶面,Co纳米粒子晶粒尺寸大约为40 nm。并且可观察到Co纳米粒子被包覆在TiB2载体里,TiB2外壳厚度大约为2.554 nm,这是SMSI的典型特征,有利于增强金属与载体之间的电子传递[27]。Co/TiB2和TiB2的XRD图如图1f所示。TiB2样品的所有衍射峰都能与TiB2的标准衍射峰相对应(PDF No.35-0741)。负载 Co纳米粒子之后,分别在 44°、51°和76°处出现3个明显的新衍射峰,对应面心立方Co的(111)、(200)和(220)晶面(PDF No.15-0806),除此之外并无其他明显杂峰。通过TiB2载体和Co/TiB2的XPS全谱图(图1g)可以看出,负载了Co纳米粒子之后,全谱中出现明显的Co2p轨道峰,进一步证明了该催化剂的成功制备。
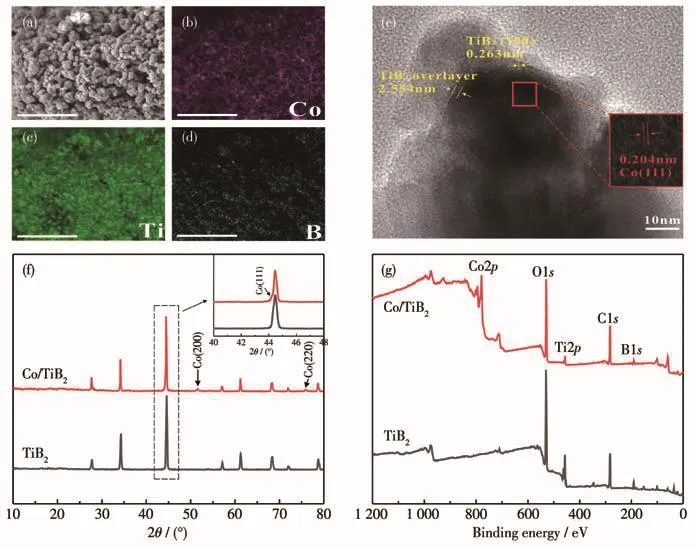
图1 (a)Co/TiB2的SEM图以及(b~d)EDS元素分布图;(e)Co/TiB2的TEM图;Co/TiB2与TiB2的(f)XRD图以及(g)XPS全谱图Fig.1 (a)SEM and(b-d)EDS elemental distributions images of Co/TiB2;(e)TEM images of Co/TiB2;(f)XRD patterns and(g)XPS full spectra of Co/TiB2 and TiB2
通过XPS精细谱进一步研究了Co纳米粒子和TiB2之间的相互作用,结果如图2所示。TiB2的Ti2p轨道的XPS谱图(图2a)显示存在Ti2+(TiB2,454.5 eV)和 Ti4+(TiO2,458.8 eV),Ti元素氧化物的检出可归因于表面钝化[28]。当Co纳米粒子沉积在TiB2上时,TiB2峰面积显著降低,这可能是由于B迁移到Co纳米粒子表面形成包覆层。此外,由于TiO2仅在TiB2表面形成,因此Co纳米粒子的沉积不会降低整体Ti4+的状态,而只会降低Ti与B之间的相互作用(由于高的Co覆盖率),这与Ti2p谱图结果一致。B1s在187.8 eV(Co/TiB2为187.9 eV)处的特征峰归属于TiB2,而 192.7 eV(Co/TiB2为 192.2 eV)处的特征峰则归因于 B2O3(图 2b)。相比于 TiB2,Co/TiB2中 TiB2对应特征峰的结合能从192.7 eV负位移到192.2 eV,并伴随着相对峰面积减小。我们认为这是由于Co纳米粒子沉积后表面TiB2和B2O3物种的损失,这与Ti2p谱图的变化一致(图2a)[27]。Co2p精细谱如图2c所示,金属Co(Co0)的相对峰强度较小,进一步证明绝大部分Co纳米粒子被TiB2载体包覆。另外,检测到Co存在+2价和+3价形式,由于Co/TiB2的XRD图(图1f)并未出现明显的CoO与Co2O3的衍射峰,因此我们推测Co2+和Co3+是未被TiB2包覆而裸露在外的那部分Co纳米粒子与空气接触后表面氧化所致。Co/TiB2的O1s谱图中(图2d),位于529.9 eV的峰归属于TiB2表面的TiO2和Co的氧化态晶格中的金属-氧键合作用,这与Co2p谱图结果一致。此外,Co/TiB2的O1s谱图中位于531.4和532.3 eV的峰分别归属于—OH和B2O3,并伴随着B2O3相对峰面积的减小,这可能是由于Co纳米粒子的沉积使得表面B2O3减少,而—OH基团的存在反映出催化剂具有良好的亲水性,促进了反应在水溶液中的催化效率[29]。

图2 Co/TiB2和TiB2的XPS谱图:(a)Ti2p3/2、(b)B1s、(c)Co2p和(d)O1sFig.2 XPS spectra of Co/TiB2 and TiB:(a)Ti2p3/2,(b)B1s,(c)Co2p,and(d)O1s
2.2 催化性能检测
2.2.1 催化产氢性能检测
Co/TiB2在空气下催化NH3BH3溶液产氢表现出优异的活性,并且无需任何形式的添加剂如NaOH等。图3a显示了不同Co负载量对催化反应的影响,较高的Co负载量会有更好的产氢活性,并且在负载量为5%以上时可达到nNH3BH∶3nH2=1∶3(150µmol),当达到15%的Co负载量时催化剂的活性达到最大值,经计算产氢速率为565.8molH2·molcat-1·h-1。随着负载量的继续增加,产氢活性逐渐降低,这是活性位点被覆盖所致。图3b是15%的Co负载到不同载体(TiB2、HfB2、ZrB2、MoB2和 TiO2)后催化NH3BH3溶液产氢图,与Co/TiB2的催化性能相比,其他催化剂在相同的反应条件下的催化活性相对较低,我们认为Co纳米粒子与TiB2之间的强相互作用增强了电子传递使得催化活性提升。图3c显示了Co/TiB2催化不同浓度NH3BH3溶液的产氢量,随着NH3BH3溶液浓度增加,产氢量也随之增加。另外观察到,NH3BH3溶液浓度在 0.05 mol·L-1时的前 30 min内,均可达到1∶3,但随着NH3BH3浓度的继续增加,最终未达到1∶3。根据NH3BH3水解产氢方程:NH3BH3+2H2O⇌NH4BO2+3H2↑,我们分析认为,在密封试管内H2产量增加的同时管内气压随之增加,平衡逐渐不利于正向进行,导致反应最终未完全进行。

图3 (a)不同Co负载量以及(b)Co负载不同载体(TiB2、HfB2、TiO2、ZrB2、MoB2)催化NH3BH3溶液(0.01 mol·L-1)产氢;(c)Co/TiB2催化不同浓度NH3BH3溶液产氢Fig.3 (a)H2 evolution from NH3BH3 aqueous solution(0.01 mol·L-1)catalyzed by TiB2 with different Co loadings and(b)Co supported on different supports(TiB2,HfB2,ZrB2,MoB2,and TiO2)catalysts;(c)H2 evolution from NH3BH3 aqueous solution with different concentrations catalyzed by Co/TiB2
2.2.2 催化降解性能检测
我们的催化产氢体系另一个特点是可以进一步串联降解有机污染物。4-NP是废水中常见的一类高毒性含氮污染物,而将其加氢氨基化还原为低毒性且更有利用价值的4-AP(药物、农药、染料和色素的关键中间体)具有重要的工业意义[23,30]。为此我们选取了4-NP作为目标物,在NH3BH3存在下进行4-NP氨基化实验。4-NP溶液本身呈淡黄色,由于NH3BH3溶液显弱碱性,根据相关文献报道,在碱性环境下4-NP会转变为4-NP离子,并且溶液的λmax会由317 nm增加到400 nm,溶液颜色逐渐变为深黄色,因此观察到混合溶液于0 min时呈深黄色(图4a插图)[31]。

图4 (a)Co/TiB2室温下催化4-NP氨基化和(b)氨基化反应后与0.1 mmol·L-1的4-AP溶液对比的UV-Vis吸收光谱图;(c)Co/TiB2室温下催化4-NP氨基化的动力学曲线(λmax=400 nm)Fig.4 UV-Vis absorption spectra of(a)4-NP amination at room temperature catalyzed by Co/TiB2 and(b)compared with 0.1 mmol·L-14-AP aqueous after amination reaction;(c)Kinetic curve of 4-NP amination at room temperature catalyzed by Co/TiB2(λmax=400 nm)
为了进一步研究催化体系对有机污染废水的降解活性,我们选取了常见的3种偶氮染料AO7、AR1和MO模拟印染废水,在NH3BH3存在下进行降解实验。由UV-Vis吸收光谱可知3种偶氮染料的λmax别为484、532和466 nm,这分别是其显色基团偶氮基(—N=N—)的吸收峰位置。
图5a、5c、5e分别是室温下Co/TiB2对AO7、AR1和MO的催化降解效果图。UV-Vis吸收光谱图中λmax处的吸收峰强度不断下降并最终消失,说明Co/TiB2对3种偶氮染料均有显著的降解效果且降解率接近100%。同时,我们注意到反应过程中混合溶液颜色逐渐变为无色透明(图5a、5c、5e的插图),这说明偶氮染料分子中的—N=N—发生了断键。由图5b、5d、5f可知偶氮染料的降解过程同样符合准一级反应动力学模型,并且降解AR1染料也存在一个诱导期,推测和4-NP氨基化反应类似,由于降解反应初期AR1阴离子和NH3BH3分子在催化剂表面未达到吸附-脱附平衡,降解反应速率常数较低。Co/TiB2催化降解偶氮染料AO7、AR1和MO的反应速率常数分别为0.34、0.26和0.31 min-1。值得注意的是,相比其他方法,该降解技术无需添加光、热等外部能量或者额外助剂(如H2O2、过一硫酸盐、过氧乙酸等)[32],室温下即可实现偶氮染料高效降解,这大大降低了偶氮染料的处理成本。
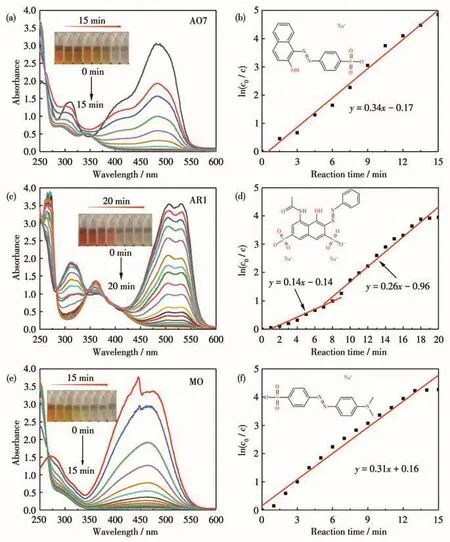
图5 Co/TiB2室温催化降解偶氮染料(a)AO7、(c)AR1和(e)MO的UV-Vis吸收光谱图以及(b、d、f)在相应染料的λmax处得到的动力学曲线Fig.5 (a)UV-Vis absorption spectra of azo dyes(a)AO7,(c)AR1,and(e)MO degraded at room temperature catalyzed by Co/TiB2,and(b,d,f)kinetic curves obtained at λmaxof the corresponding dyes
催化剂在使用过程中的循环性能至关重要,为此我们进行了循环降解实验来评估催化剂的稳定性。选用AO7染料作为目标有机污染物进行了7次循环实验,结果如图6a所示。在第1次使用时,15 min内AO7染料的降解率接近100%,并且经过4次循环后仍有93%以上的降解率,而从第5次循环开始降解率有所下降,我们认为主要是催化剂在多次循环过程中的部分损失导致降解率下降。图6b显示了Co/TiB2的磁滞回线,磁滞环是关于原点对称的非线性磁滞回线,表明催化剂在室温下表现出了良好的磁性特征,饱和磁化强度和矫顽力分别为12.73 emu·g-1和400.2 G。强磁性特征使得催化剂在外加磁场作用下80 s内即可从溶液中分离,经过4次重复实验仍能达到95%以上的磁回收率(表1),达到便于回收利用和避免对水体造成二次污染的效果,如图6b插图所示。

表1 Co/TiB2的磁回收率Table 1 Magnetic recovery rate of Co/TiB2

图6 (a)Co/TiB2催化降解AO7的循环实验;(b)Co/TiB2的磁滞回线Fig.6 (a)Cycling experiment for AO7 degradation catalyzed by Co/TiB2;(b)Magnetic hysteresis loop of Co/TiB2
2.3 反应路径及机理
为了进一步说明催化降解有机污染物可能的反应过程,我们以DMPO为自由基捕捉剂,选用AO7染料作为目标有机污染物进行了EPR光谱分析,结果如图7a所示。当体系中不加入NH3BH3时,并没有检测到明显的自由基信号,说明此时AO7染料仅仅是在Co/TiB2表面吸附而未发生催化降解反应(图7a黑线)。加入NH3BH3后检测到明显的信号并可归属于DMPO-H加合物,证明了氢自由基(·H)的存在。我们认为·H来源于NH3BH3中B—H键断裂,而Co/TiB2加速了·H生成[31]。另外可注意到,加入AO7染料后相比于无添加的EPR光谱对应峰强度有所减弱,我们认为这是由于·H降解有机污染物而被消耗。为了探究·H在反应过程中及反应后的行为表现,我们利用GC-TCD气相色谱仪对加入有机污染物的催化体系产氢性能进行检测,结果如图7b所示。可以看出,在相同时间下,加入AO7染料的产氢量相比于无添加有明显降低,并且图7b插图显示加入AO7染料的体系在2 min后褪为无色透明,说明此时·H主要作用于降解有机污染物,并且在2 min之后产氢速率呈上升趋势;对此我们分析,当体系中有机污染物不存在或者已经降解完后,生成的·H会在催化剂表面结合生成H2分子;而当有机污染物存在时,·H会更优先降解有机污染物。
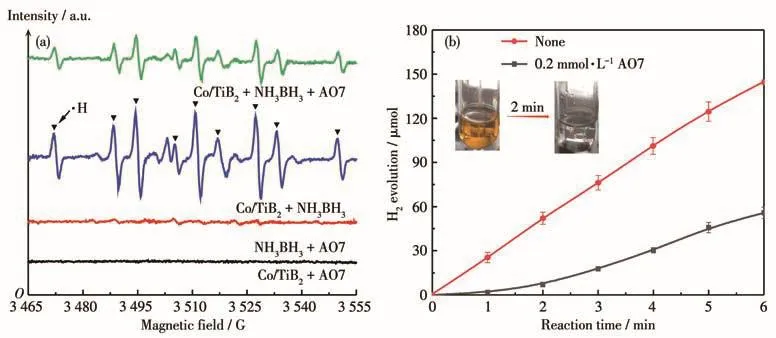
图7 (a)Co/TiB2+AO7、NH3BH3+AO7、Co/TiB2+NH3BH3和Co/TiB2+NH3BH3+AO7体系的EPR光谱;(b)有无AO7时Co/TiB2催化NH3BH3溶液(0.01 mol·L-1)产氢Fig.7 (a)EPR spectra of the systems of Co/TiB2+AO7,NH3BH3+AO7,Co/TiB2+NH3BH3,and Co/TiB2+NH3BH3+AO7;(b)H2 evolution from NH3BH3 aqueous solution(0.01 mol·L-1)catalyzed by Co/TiB2 with and without AO7
基于以上结果,我们推测了催化降解有机污染物可能的反应路径,如图8所示。首先NH3BH3和有机污染物分子共同吸附在Co/TIB2表面以保证电荷转移,之后NH3BH3催化水解产生·H以还原有机污染物,其中4-NP被加氢氨基化还原为4-AP;偶氮染料中的—N=N—被·H先初步还原为肼衍生物(—HN—NH—),接着·H进一步还原使N—N键断开生成不同小分子产物[23]。
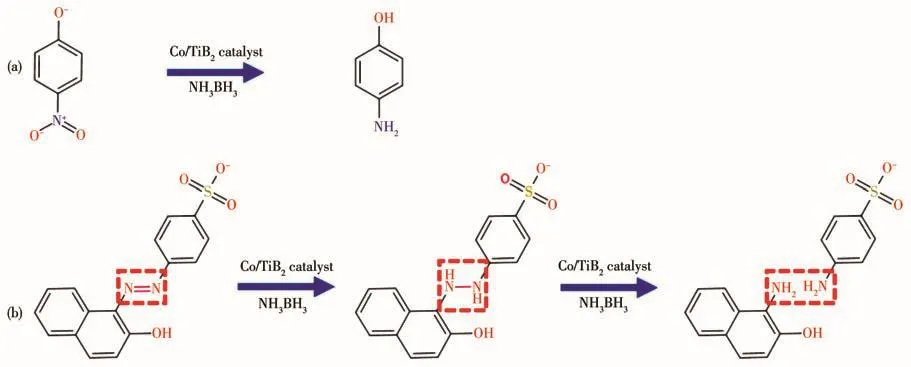
图8 有机污染物的催化还原过程:(a)4-NP氨基化过程;(b)偶氮染料降解过程Fig.8 Catalytic reduction process of organic pollutants:(a)4-NP amination process;(b)azo dye degradation process
3 结论
通过简单的沉淀-沉积法成功制备了Co/TiB2磁性纳米催化剂,并应用于室温催化NH3BH3溶液产氢。相比于ZrB2、MoB2等载体,Co纳米粒子被TiB2包覆形成金属-载体强相互作用,增强了彼此间的电子传递,使其表现出优异的催化产氢活性,产氢速 率高 达 565.8molH2·molcat-1·h-1。 除此 之 外 ,Co/TiB2+NH3BH3催化体系在不添加任何外部能量或者额外助剂下即可高效串联降解有机污染物,7 min内催化4-NP氨基化的转化率接近100%,氨基化反应速率常数高达0.72 min-1;模拟印染废水中,降解AO7这一偶氮染料的反应速率常数达到0.34 min-1。通过EPR-DMPO自由基捕获实验检测出Co/TiB2+NH3BH3催化体系产生大量的活性还原物种·H,·H在Co/TiB2表面结合生成H2分子;并且得益于·H的强还原性,有机污染物能够被高效降解。Co/TiB2在室温下良好的磁性特征使其通过外加磁场即可快速回收利用,避免催化剂对水体的二次污染。本串联催化技术为开发高性能、低成本、节能环保的制氢材料和处理有害有机污染物提供了新的思路和方法。
猜你喜欢
纺织标准与质量(2022年3期)2022-08-10
家庭医药(2021年8期)2021-07-28
煤气与热力(2021年2期)2021-03-19
父母必读(2021年3期)2021-02-04
河南科学(2020年7期)2020-09-10
化工设计(2020年2期)2020-05-01
陕西科技大学学报(2018年1期)2018-01-11
分析化学(2017年12期)2017-12-25
郑州大学学报(理学版)(2017年1期)2017-04-07
中国新技术新产品(2017年3期)2017-03-07
