
LncRNA MALAT1调节miR-383-5p/SOCS3轴对人牙周膜干细胞的影响
2022-12-02 05:01:28顾婷立刘亚华
口腔医学 2022年11期
顾婷立,刘亚华,钱 靓,章 茜
牙周炎是一种牙周支持组织的慢性感染性疾病[1]。在炎症条件下,人牙周膜干细胞(human periodontal ligament stem cells,hPDLSCs)成骨分化能力降低,无法实现有效的牙周支持组织再生[2]。长链非编码RNA(long non-coding RNA,LncRNA)参与多种生物过程[3],肺腺癌转移相关转录本1(metastasis-associated lung adenocarcinoma transcript 1,MALAT1)是一种与牙周炎进展有关的LncRNA。研究显示MALAT1在牙周炎患者的hPDLSCs中明显高表达[4],可促进人牙龈成纤维细胞中炎性因子的分泌[5],但MALAT1在牙周炎中的分子机制尚不清楚。既往研究表明,微小RNA(microRNA,miRNA)可以通过干扰牙周细胞的分化、再生和修复来调节牙齿发育网络[6-7]。微小RNA-383-5p(miRNA-383-5p,miR-383-5p)可参与调节hPDLSCs的成骨分化[8]。此外,细胞因子信号转导负调控因子3(suppressor of cytokine signaling 3,SOCS3)在多种细胞生物学过程中发挥重要作用,已被证实与糖尿病小鼠的牙周炎症有关[9-10]。本研究首次探讨了MALAT1/miR-383-5p/SOCS3轴在hPDLSCs增殖、凋亡以及成骨分化中的作用及LncRNA-miRNA-mRNA调控网络,为牙周修复提供新的治疗靶点。
1 材料与方法
1.1 材料与仪器
hPDLSCs(PC-025h)(赛奥斯,中国武汉),牙龈卟啉单胞菌脂多糖(lipopolysaccharide,LPS)(SMB00610)(Sigma,美国),青霉素-链霉素-谷氨酰胺(10378016)、胎牛血清(1009914C)和DEME培养基(11965118)(Gibco,美国),PrimeScript cDNA合成试剂盒(D6110A)、Green Premix Ex Taq(RR420A)(TAKARA,日本),一抗SOCS3(ab280884)、GAPDH(ab8245)、PCNA(ab29)、Ki-67(ab92742)、Bcl-2(ab32124)、Bax(ab32503)、OSX(ab209484)和OCN(ab133612)(Abcam,英国),Immun-StarTMHRP化学发光试剂盒(1705040)(Bio-Rad,美国),CCK-8试剂盒(CA1210)(Solarbio,中国),TUNEL凋亡试剂盒(AT2190)、BCIP/NBT显色试剂盒(APR1100)、茜素红S(alizarin red S,ARS)染色液(A39470)(吉至,中国上海),成骨诱导试剂盒(FY200006)(弗元生物,中国上海),碱性磷酸酶(alkaline phosphatase,ALP)活性测定试剂盒(mlE3570)(酶联生物,中国上海),TNF-α ELISA试剂盒(EK0525)、IL-6 ELISA试剂盒(EK0410)和IL-1β ELISA试剂盒(EK0392)(博士德,中国武汉),双荧光素酶检测试剂盒(LF001)(Gene Copoeia,美国)。
SpectraMax iD3型多功能酶标仪(Molecular Devices,美国),7500实时荧光定量PCR仪器(ABI,美国),OmegaLum C 化学发光凝胶成像系统(aplegen,美国)、DMi8荧光显微镜(Leica,德国)。
1.2 细胞分组与转染
取第3~5代hPDLSCs用于实验,将细胞按以下分组处理。Control组:不做处理;LPS组:仅用LPS处理;LPS+si-NC组:转染si-NC后LPS处理;LPS+si-MALAT1组:转染靶向MALAT1的小干扰RNA(si-MALAT1)后LPS处理;LPS+si-MALAT1+in-miR-383-5p组:转染si-MALAT1和miR-383-5p抑制物(in-miR-383-5p)后LPS处理;LPS+si-MALAT1+SOCS3组:转染si-MALAT1和SOCS3过表达质粒(SOCS3)后LPS处理。LPS终浓度1 mg/L[11],所有细胞处理48 h后用于后续实验。
1.3 ELISA实验
收集各组hPDLSCs细胞上清液,按照试剂盒说明书检测TNF-α、IL-6和IL-1β水平。酶标仪测定各孔450 nm处OD值,根据标准曲线获得相应的细胞因子水平。
1.4 RT-qPCR实验
提取各组hPDLSCs总RNA,使用PrimeScript cDNA合成试剂盒制备cDNA。随后使用Green Premix Ex Taq试剂进行RT-qPCR,引物序列见表1。使用2-ΔΔct法归一化为GAPDH/U6表达。

表1 引物序列表
1.5 Western blot实验
使用RIPA裂解缓冲液提取各组hPDLSCs总蛋白,经定量后通过适当的SDS-PAGE分离蛋白(50 μg/泳道)并转移到PVDF膜上,然后使用5%脱脂牛奶在TBST缓冲液中封闭2 h。将膜与SOCS3、PCNA、Ki-67、Bcl-2、Bax、OSX、OCN和GAPDH一抗(1∶1 000)4 ℃过夜孵育。一抗孵育后使用HRP偶联的二抗(1∶1 000)在室温下培养45 min。经Immun-StarTMHRP化学发光试剂显影后于检测器上观察蛋白质条带。灰度值使用Image J量化。
1.6 CCK-8检测
将hPDLSCs细胞(1×104个/孔)接种在96孔板中,培养24 h后每孔加入10 μL的CCK-8试剂,4 h后再使用酶标仪测量450 nm处OD值。
1.7 TUNEL检测
将hPDLSCs细胞在室温下用4%多聚甲醛固定30 min后,使用0.2% Triton X-100透化8 min。加入TUNEL溶液在37 ℃下孵育60 min,PBS洗涤后再加入DAPI染色液对细胞核染色8 min。使用荧光显微镜观察6个视野中的凋亡细胞,凋亡指数=凋亡细胞数(绿色荧光)÷总细胞数(蓝色荧光)×100%。
1.8 ALP染色和活性分析
将转染的hPDLSCs细胞接种在6孔培养板中并在成骨诱导培养基中培养14 d后,①ALP活性分析:收集细胞后按照ALP活性测定试剂盒说明书操作,在微孔板分光光度计520 nm波长处检测细胞的OD值;②ALP染色:PBS洗涤细胞后使用4%多聚甲醛固定30 min,向每孔添加适量的BCIP/NBT溶液避光孵育后进行拍照。
1.9 ARS染色
将转染的hPDLSCs细胞接种在6孔培养板中并在成骨诱导培养基中培养21 d后,PBS洗涤后加入4%多聚甲醛固定30 min,加入ARS染色溶液染色30 min。使用倒置显微镜对矿化结节进行拍照,酶标仪于520 nm波长下检测OD值。
1.10 双荧光素酶报告基因检测
使用Starbase数据库确定MALAT1和miR-383-5p之间的结合位点,并预测miR-383-5p的潜在靶标SOCS3。构建含有预测miR-383-5p结合位点的野生型MALAT1/3′-UTR-SOCS3(WT-MALAT1/WT-3′-UTR-SOCS3)和突变型MALAT1/3′-UTR-SOCS3(MUT-MALAT1/MUT-3′-UTR-SOCS3),并克隆到psiCHECK-2表达载体上,使用Lipofectamine 3000转染试剂将WT-MALAT1/3′-UTR-SOCS3或MUT-MALAT1/3′-UTR-SOCS3和miR-383-5p mimics或miR-NC共转染到hPDLSCs细胞中。48 h后根据试剂盒说明分析荧光素酶活性。
1.11 统计学分析
使用SPSS 22.0分析数据,并在本研究中以平均值±标准差表示。两组之间比较采用独立t检验,三组或三组以上的差异比较采用单因素方差分析和Turkey′s事后检验。P<0.05被认为差异具有统计学意义。
2 结 果
2.1 hPDLSCs细胞中炎症因子表达情况
表2显示,与Control组比较,LPS组hPDLSCs细胞中TNF-α、IL-1β和IL-6水平明显升高(P<0.05)。与LPS+si-NC组比较,LPS+si-MALAT1组hPDLSCs细胞中TNF-α、IL-1β和IL-6水平明显降低(P<0.05)。与LPS+si-MALAT1组比较,LPS+si-MALAT1+in-miR-383-5p组和LPS+si-MALAT1+SOCS3组hPDLSCs细胞中TNF-α、IL-1β和IL-6水平均明显升高(P<0.05)。

表2 各组hPDLSCs细胞中TNF-α、IL-1β和IL-6水平
2.2 hPDLSCs细胞中MALAT1、miR-383-5p和SOCS3表达情况
与Control组比较,LPS组hPDLSCs细胞中MALAT1和SOCS3表达水平均升高,miR-383-5p表达水平降低(P<0.05)。与LPS+si-NC组比较,LPS+si-MALAT1组hPDLSCs细胞中MALAT1和SOCS3表达水平均降低,miR-383-5p表达水平升高(P<0.05)。与LPS+si-MALAT1组比较,LPS+si-MALAT1+in-miR-383-5p组hPDLSCs细胞中MALAT1表达水平变化无差异(P>0.05),miR-383-5p表达水平降低,SOCS3表达水平升高(P<0.05);LPS+si-MALAT1+SOCS3组hPDLSCs细胞中MALAT1和miR-383-5p表达水平变化无差异(P>0.05),SOCS3表达水平升高(P<0.05)(图1、表3)。
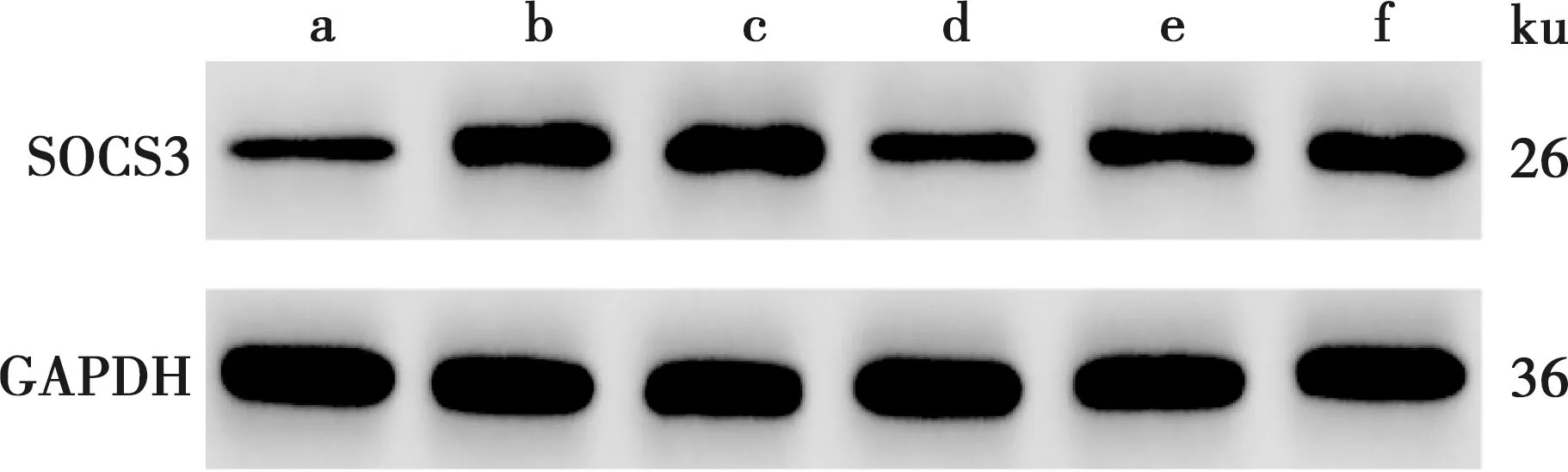
a~f依次为Control组、LPS组、LPS+si-NC组、LPS+si-MALAT1组、LPS+si-MALAT1+in-miR-383-5p组和LPS+si-MALAT1+SOCS3组
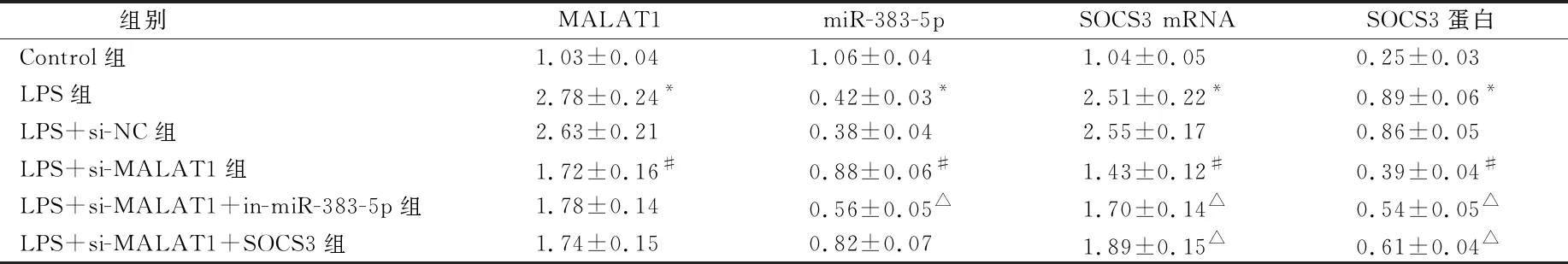
表3 各组hPDLSCs细胞中MALAT1、miR-383-5p和SOCS3表达
2.3 hPDLSCs细胞增殖活力、凋亡情况
与Control组比较,LPS组hPDLSCs细胞活力降低,凋亡指数升高;细胞中PCNA、Ki-67、Bcl-2蛋白水平降低,Bax蛋白水平升高(P<0.05)。与LPS+si-NC组比较,LPS+si-MALAT1组hPDLSCs细胞活力升高,凋亡指数降低;细胞中PCNA、Ki-67、Bcl-2蛋白水平升高,Bax蛋白水平降低(P<0.05)。与LPS+si-MALAT1组比较,LPS+si-MALAT1+in-miR-383-5p组和LPS+si-MALAT1+SOCS3组hPDLSCs细胞活力降低,凋亡指数升高;细胞中PCNA、Ki-67、Bcl-2蛋白水平降低,Bax蛋白水平升高(P<0.05)(表4、图2~3)。
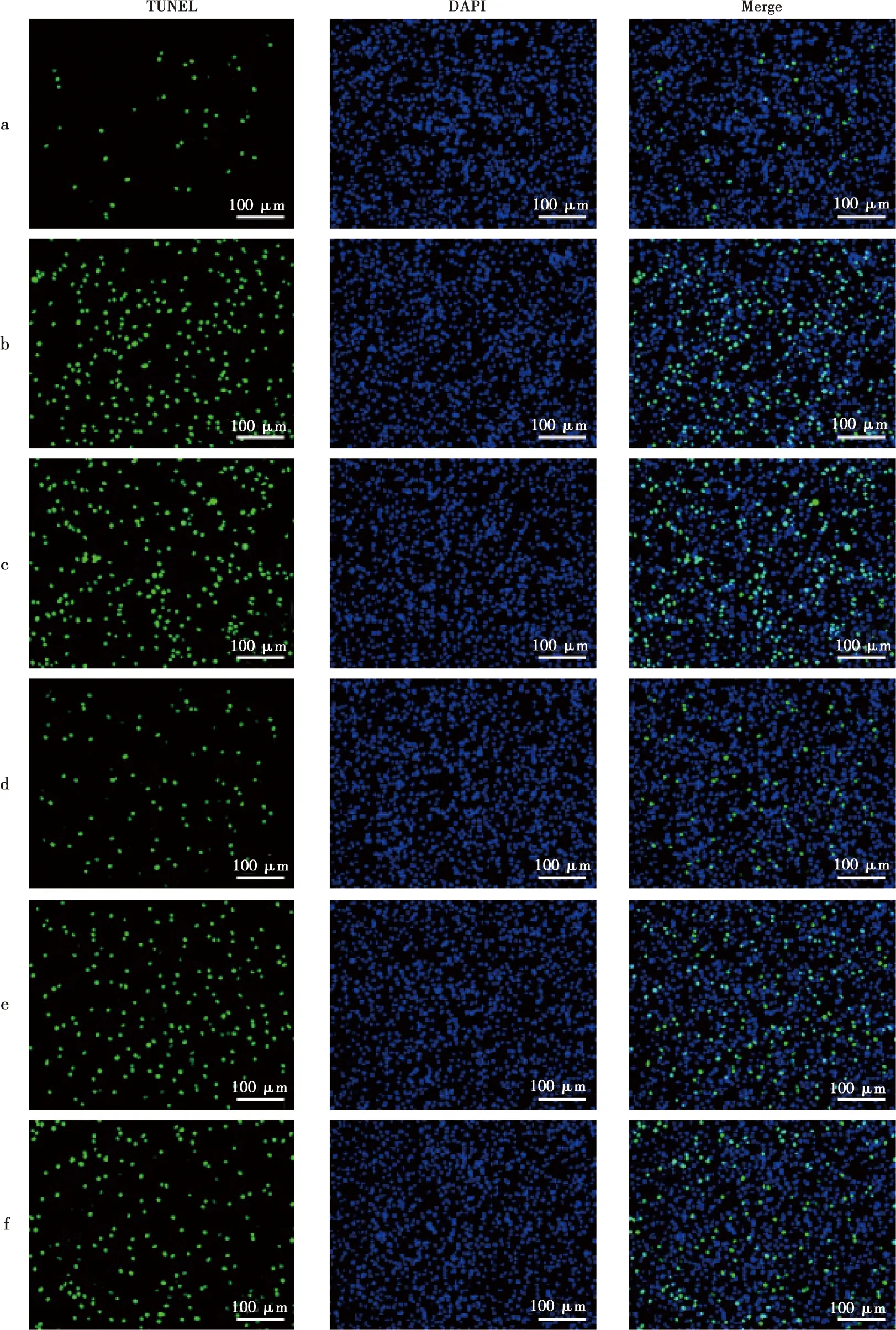
a~f依次为Control组、LPS组、LPS+si-NC组、LPS+si-MALAT1组、LPS+si-MALAT1+in-miR-383-5p组和LPS+si-MALAT1+SOCS3组
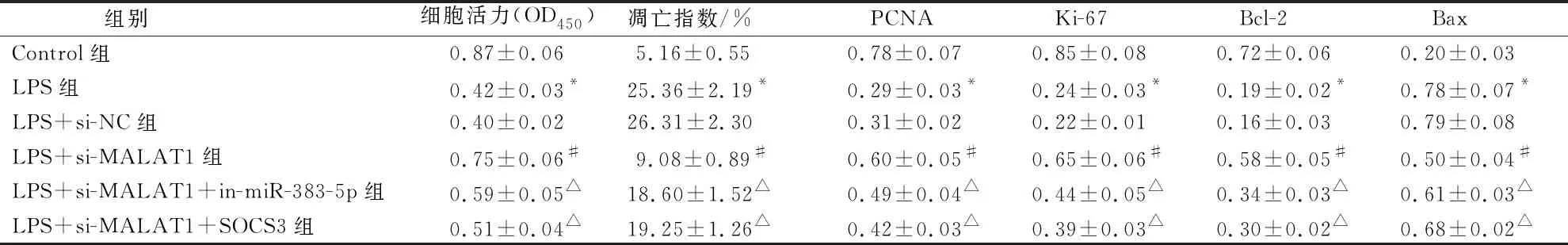
表4 各组hPDLSCs细胞活力、凋亡指数以及PCNA、Ki-67、Bcl-2和Bax蛋白水平统计
2.4 hPDLSCs细胞成骨分化情况
与Control组比较,LPS组hPDLSCs细胞中ALP染色和活性、ARS钙沉积物的浓度减少,细胞中OSX和OCN蛋白水平降低(P<0.05)。与LPS+si-NC组比较,LPS+si-MALAT1组hPDLSCs细胞中ALP染色和活性、ARS钙沉积物的浓度升高,细胞中OSX和OCN蛋白水平升高(P<0.05)。与LPS+si-MALAT1组比较,LPS+si-MALAT1+in-miR-383-5p组和LPS+si-MALAT1+SOCS3组hPDLSCs细胞中ALP染色和活性、ARS钙沉积物的浓度减少,细胞中OSX和OCN蛋白水平降低(P<0.05)(表5、图4~5)。
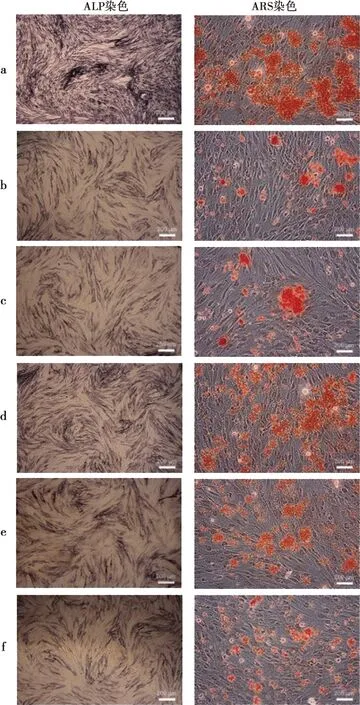
a~f依次为Control组、LPS组、LPS+si-NC组、LPS+si-MALAT1组、LPS+si-MALAT1+in-miR-383-5p组和LPS+si-MALAT1+SOCS3组
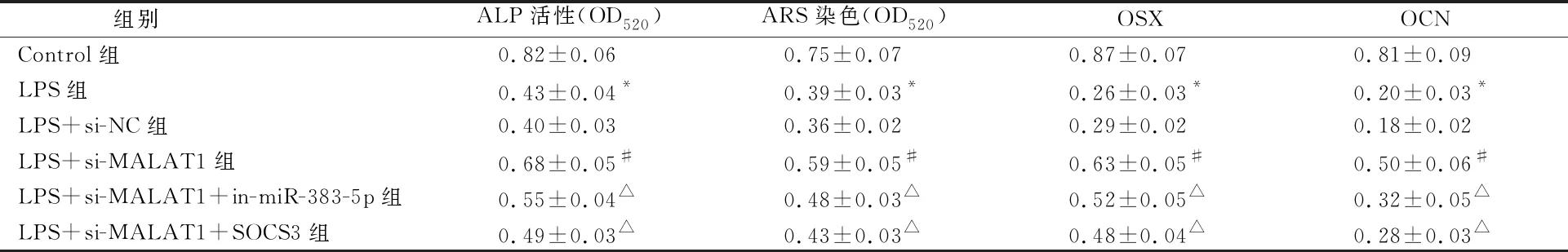
表5 各组hPDLSCs细胞中ALP活性、ARS染色和OSX、OCN蛋白水平统计

a~f依次为Control组、LPS组、LPS+si-NC组、LPS+si-MALAT1组、LPS+si-MALAT1+in-miR-383-5p组和LPS+si-MALAT1+SOCS3组
2.5 MALAT1、miR-383-5p和SOCS3在hPDLSCs细胞中的关系
miR-383-5p与MALAT1或SOCS3 3′-UTR存在靶向结合位点(图6)。双荧光素酶结果显示(表6),转染miR-383-5p mimics能特异性降低WT-MALAT1或WT-3′-UTR-SOCS3的荧光素酶活性(P<0.05),而不能特异性降低MUT-MALAT1或MUT-3′-UTR-SOCS3的荧光素酶活性(P>0.05);同时转染miR-NC对WT-MALAT1或WT-3′-UTR-SOCS3与MUT-MALAT1或MUT-3′-UTR-SOCS3荧光素酶活性均无影响(P>0.05)。因此可得出,在hPDLSCs细胞中MALAT1与miR-383-5p之间存在相互作用关系,miR-383-5p与SOCS3之间存在相互作用关系。

表6 双荧光素酶报告基因测定miR-383-5p与MALAT1或SOCS3 3′-UTR靶向关系
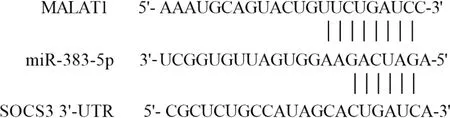
图6 Starbase网站预测miR-383-5p与MALAT1或SOCS3 3′-UTR互补位点
3 讨 论
牙周炎是一种慢性疾病,药物治疗可作为机械清除菌斑牙石的辅助治疗[12]。hPDLSCs因具有高增殖、自我更新和多分化能力,在牙周炎组织修复中至关重要[2]。本研究通过LPS构建hPDLSCs细胞炎症模型发现,LPS刺激后hPDLSCs表达促炎因子(TNF-α、IL-1β和IL-6)水平升高,与Nie等[11]报道一致。LPS还可通过抑制软骨细胞增殖相关蛋白(PCNA、Ki-67)表达,同时上调促凋亡蛋白Bax,下调抗凋亡蛋白Bcl-2水平,进而抑制细胞增殖,诱导细胞凋亡[13],本研究结果与之相似。ALP活性和矿化程度是成骨分化的标志[14]。本研究中LPS刺激后hPDLSCs中ALP的活性和矿化程度均受到抑制,同时成骨分化标志物OCN和转录因子OSX的表达下降。以上数据表明,LPS刺激在诱导hPDLSCs炎症和凋亡损伤的同时也削弱了细胞增殖和成骨分化能力。
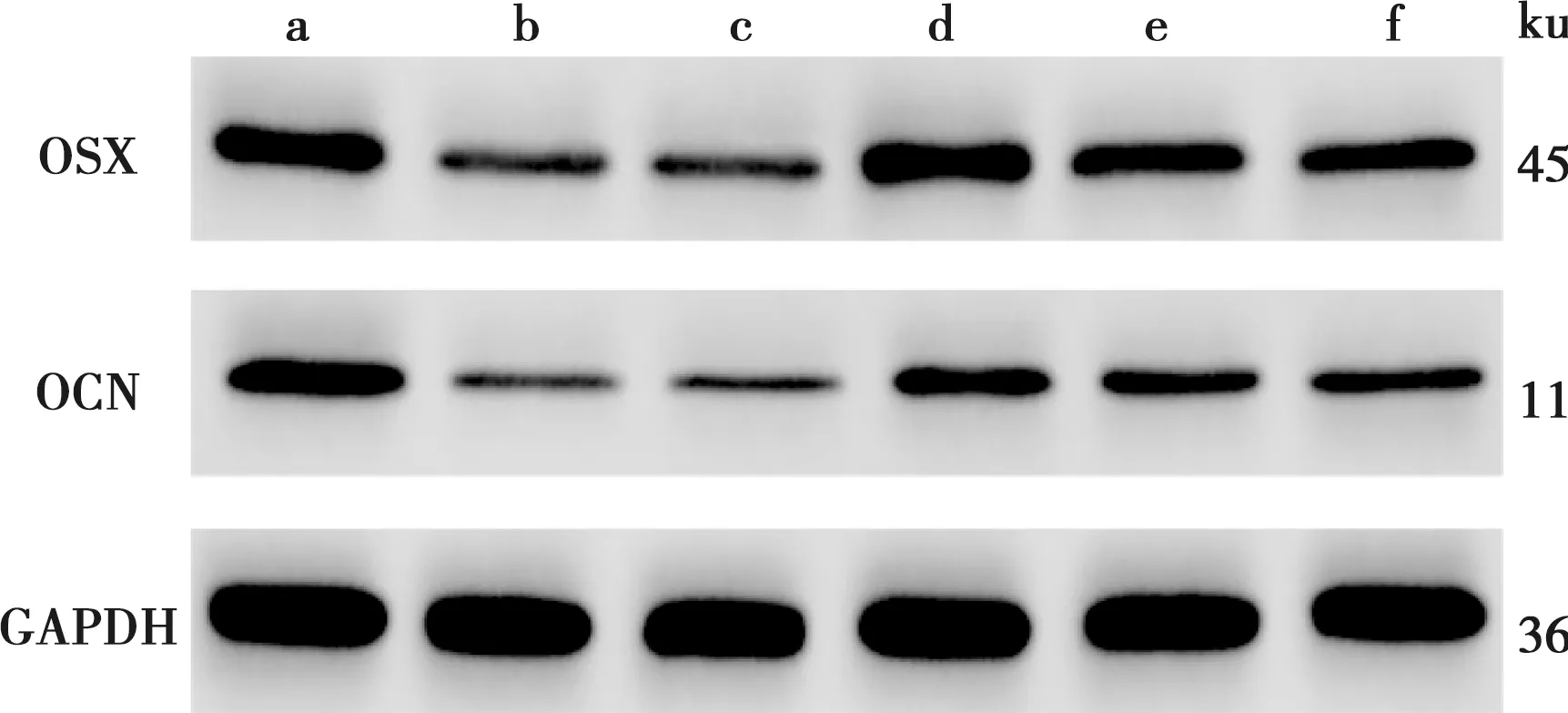
a~f依次为Control组、LPS组、LPS+si-NC组、LPS+si-MALAT1组、LPS+si-MALAT1+in-miR-383-5p组和LPS+si-MALAT1+SOCS3组
MALAT1高表达可促进帕金森病等疾病的发展[15],下调MALAT1表达可能有利于治疗疾病。MALAT1通过上调成纤维细胞生长因子2促进hPDLSCs的增殖[4]。此外,MALAT1还可通过海绵化miR-20a和激活TLR4促进LPS刺激的人牙龈成纤维细胞中炎性细胞因子的产生[5]。MALAT1敲低后可通过miR-769-5p/HIF3A轴削弱LPS刺激下hPDLSCs表达促炎因子(TNF-α、IL-1β和IL-6)的水平,进而抑制炎症反应[16]。本研究结果发现,LPS刺激可明显上调hPDLSCs中MALAT1的表达,进一步结果表明敲低MALAT1不仅抑制了LPS刺激的hPDLSCs促炎因子(TNF-α、IL-1β和IL-6)的分泌,同时增强了细胞活力并抑制了细胞凋亡。此外,MALAT1敲低部分恢复了LPS刺激下hPDLSCs下调的成骨分化能力。以上这些结果表明敲低MALAT1可能抑制牙周炎的发生和发展。
LncRNA通过海绵化miRNA并调控靶基因的表达,进而参与调控多个生物过程,如LncRNA HOTAIRM1海绵化miR-433-5p促进PIK3CD表达,进而加速多囊卵巢综合征的发展[17]。越来越多的研究表明,MALAT1可以作为一种内源竞争RNA来调节miRNA的表达和靶基因的活性[16]。通过使用生物信息学对MALAT1预测靶点进行分析,确定了很多潜在的靶点包括miR-383-5p、miR-4524b-5p、miR-503-5p等,这些靶点多与人类癌症的发展相关。其中,miR-383-5p可通过抑制HDAC9表达促进hPDLSCs的成骨分化[8]。以往研究表明,miR-383-5p对前列腺癌等多种癌症的生长具有抑制作用[18]。因此,本研究选择miR-383-5p进行机制研究。有研究表明,MALAT1下调后可通过与miR-383-5p直接作用来调节人脐静脉内皮细胞增殖、凋亡、迁移和侵袭[19]。本研究发现,MALAT1也可以通过与hPDLSCs中miR-383-5p结合发挥作用,下调MALAT1表达可明显上调miR-383-5p表达。此外,LPS刺激后,hPDLSCs中miR-383-5p表达降低,提示MALAT1可能通过miR-383-5p参与牙周炎发展过程。进一步研究发现,抑制miR-383-5p表达可部分减弱MALAT1敲低对hPDLSCs的影响,表明MALAT1可能通过海绵化miR-383-5p对牙周炎产生影响。miR-383-5p的靶基因是SOCS3。脱氢丁香酚可通过抑制STAT3/SOCS3通路改善LPS诱导的哮喘模型肺部炎症[20];SOCS3还被证实可作为骨髓间充质干细胞成骨分化和骨形成的负调节因子[21]。Wang等[10]研究证实,降低SOCS3的表达可抑制衰老相关分泌表型,进而抑制糖尿病小鼠牙周炎症的发展。本研究结果显示,LPS诱导的hPDLSCs中SOCS3表达升高,且miR-383-5p可靶向调控SOCS3表达,而过表达SOCS3可部分逆转MALAT1敲低后对hPDLSCs细胞的影响,证实了下调MALAT1通过调控miR-383-5p/SOCS3轴减弱LPS诱导的hPDLSCs炎症损伤。
综上,本研究首次提供证据表明,下调MALAT1通过miR-383-5p减少SOCS3表达,抑制LPS诱导的hPDLSCs炎症和凋亡,促进hPDLSCs增殖和成骨分化。这可能为未来牙周炎靶向治疗方案的发展提供新的实验依据。然而,本研究仅在细胞层面对MALAT/miR-383-5p/SOCS3机制进行了探究,仍需在体内进一步研究验证;其次,SOCS3如何在hPDLSCs中发挥作用仍有待进一步探索。
猜你喜欢
口腔医学(2021年10期)2021-12-02 02:08:00
天津医科大学学报(2021年4期)2021-08-21 02:14:50
昆明医科大学学报(2021年6期)2021-07-31 07:40:38
中日友好医院学报(2021年1期)2021-04-14 01:58:32
山东医药(2020年9期)2020-05-20 01:12:16
实用口腔医学杂志(2017年6期)2017-09-19 02:51:32
中华老年口腔医学杂志(2016年2期)2017-01-15 14:24:47
中国继续医学教育(2015年4期)2016-01-07 07:38:01
中国病理生理杂志(2015年8期)2015-12-21 12:38:14
天津护理(2015年4期)2015-11-10 06:11:41
