
苗药羊耳菊中7个抗炎活性成分在Raw264.7细胞中含量测定方法的建立
2022-11-30 04:38:32周杰薛寸张青陈思颖陈艺黄静李月婷黄勇郑林巩仔鹏
江苏大学学报(医学版) 2022年6期
周杰, 薛寸, 张青, 陈思颖, 陈艺, 黄静, 李月婷, 黄勇, 郑林, 巩仔鹏
(贵州医科大学药学院,贵州省药物制剂重点实验室,省部共建药用植物功效与利用国家重点实验室,民族药与中药开发应用教育部工程研究中心,贵州 贵阳 550004)
对于药物靶点和关键药物代谢酶位于细胞内的药物而言,测定药物在细胞中浓度较其血浆中的药物浓度更有意义。当药物进入到细胞后,如何准确测定细胞内药物的含量,是开展药物在细胞内的药代动力学的前提。因此,建立快速测量和预测细胞内游离药物浓度对研究药物在细胞内的吸收、分布和代谢过程显得尤为重要[1]。虽然多成分协同发挥作用是中药民族药治疗疾病的特点,但因其所含化学成分众多,给其含量成分的测定带来巨大挑战。超高效液相色谱与质谱联用仪(ultra performance liquid chromatography tandem mass spectrometry,UPLC-MS/MS)技术具有分析速度快、分离能力强、灵敏度高和自动化程度高等特点,对于中药民族药多组分的同时测定及细胞样本中微量药物含量的定量分析均具有很大的优势[2]。
羊耳菊为菊科旋覆花属植物羊耳菊Inulacappa(Buch. -Ham. ex D. Don) DC.的新鲜或干燥全草,又名白牛胆、大力王、叶下白,是贵州云南等地少数民族常用药材,对于感冒发热、咽喉肿痛、风湿疼痛、痈疮疔毒、乳痈等症状有独特疗效[3]。近年来对羊耳菊的化学成分研究报道较多,其主要成分有酚酸类、萜类、黄酮类和挥发油等[4-7],研究发现,从羊耳菊药材中分离得到的木犀草苷(luteolin,LUT)、绿原酸(chlorogenic acid,CA)、新绿原酸(neochlorogenic acid,NCA)、隐绿原酸(cryptochlorogenic acid,CCA)、3,4-二咖啡酰基奎宁酸(3,4-dicaffeoylquinic acid,3,4-DCQA)、3,5-二咖啡酰基奎宁酸(3,5-dicaffeoylquinic acid,3,5-DCQA)、4,5-二咖啡酰基奎宁酸(4,5-dicaffeoylquinic acid,4,5-DCQA)具有抗炎活性[8-9]。羊耳菊的化学成分众多且复杂,而目前关于同时测定细胞中羊耳菊抗炎活性成分含量的方法学研究未见报道,限制了其细胞药代动力学的研究。因此,本文以小鼠巨噬细胞Raw 264.7为研究对象,采用超高液相色谱-三重四级杆质谱联用仪(UPLC-MS/MS)建立同时测定Raw 264.7细胞中LUT、CA、NCA、CCA、3,4-DCQA、3,5-DCQA、4,5-DCQA等7个成分含量的方法学,包括专属性、线性、基质效应、回收率、准确度、精密度和稳定性等,为后续进一步研究苗药羊耳菊抗炎活性成分的细胞药代动力学研究奠定基础。
1 材料与方法
1.1 材料
1.1.1 仪器 UPLC Xevo TQ-S型超高效液相三重四级杆质谱联用仪(ACQUITY UPLC I-Class系统,MassLynx质谱工作站,美国沃特世公司);CO2培养箱、超低温冰箱(美国Thermo Fisher Scientific 公司);Model 680酶标仪(伯乐生命医学产品有限公司);TS-100 F倒置显微镜(日本Nikon公司);超净工作台、Allegra 32 R台式冷冻离心机、Allegra X-30 R低温高速离心机(美国Beckman公司);高压蒸汽灭菌锅、电热恒温培养箱(上海捷呈实验仪器有限公司);KQ-500 DE 数控超声波清洗器(昆山市超声仪器有限公司);VX-Ⅲ多管涡旋振荡器(藤锦(北京)医药科技有限公司);XYN-15LP氮吹仪氮气发生器(上海析友分析仪器有限公司);EL204万分之一电子天平(上海Mettler Toledo公司);单通道微量、多通道微量移液枪(德国Eppendorf公司);电动移液枪(德国Sartorius公司);Water purifier实验室专用超纯水机(四川沃特尔水处理设备有限公司);25 cm2细胞培养瓶、细胞培养板6孔板、96孔板(无锡耐思生物科技有限公司);10、25 mL移液管(清风生化科技有限公司)。
1.1.2 试剂与药品 澳洲胎牛血清、DMEM、双抗、胰酶消化液均购于美国Gibco公司;PBS、BCA 蛋白定量试剂盒,均购于北京索莱宝科技有限公司;MTS购于普洛麦格(北京)生物技术有限公司;质谱级甲醇、乙腈(德国Merck 公司);质谱级甲酸(美国Thermo Fisher Scientific);屈臣氏饮用水(广州屈臣氏食品饮料有限公司)。
对照品:葛根素(中国食品药品检定研究院,批号110752-201514,纯度≥95.5%);绿原酸(成都埃法生物科技有限公司,批号AF8112791,纯度≥98%);隐绿原酸(成都埃法生物科技有限公司,批号AF20032707,纯度≥98%);新绿原酸(成都埃法生物科技有限公司,批号AF20030804,纯度≥98%);木犀草苷(四川维克奇生物科技有限公司,批号wkq20031803,纯度≥98%);3,4-二咖啡酰基奎宁酸(成都埃法生物科技有限公司,批号AF9060515,纯度≥98%);3,5-二咖啡酰基奎宁酸(成都埃法生物科技有限公司,批号AF20020302,纯度≥98%);4,5-二咖啡酰基奎宁酸(成都埃法生物科技有限公司,批号AF0011701,纯度≥98%)。
1.1.3 药材来源 羊耳菊药材Inulacappa(产地为广西桂林)经过贵州医科大学生药学教研室刘春花副教授鉴定为菊科旋覆花属植物羊耳菊的干燥全草。
1.1.4 实验细胞 本实验所用小鼠单核巨噬细胞RAW264.7细胞株购于武汉普诺生命科技有限公司(批号CL-0190)。
1.2 实验方法
1.2.1 羊耳菊提取物的制备方法 称取羊耳菊药材约20 kg,加10倍量60%乙醇浸泡过夜,回流提取3次,时间每次1 h,3次回流完成后,合并3次滤液,减压浓缩,得浓缩液约10 L,浓缩液经D101型大孔树脂吸附,径高比1 ∶4,吸附充分后,加水洗脱至流出液无颜色后,换60%乙醇进行洗脱,收集洗脱液,水浴蒸干,得浸膏样提取物,后续采用微波真空干燥,即得。计算得膏率为3.15%,经UPLC-MS/MS含量测定,羊耳菊提取物中木犀草苷、绿原酸、新绿原酸、隐绿原酸、3,4二咖啡酰基奎宁酸、3,5二咖啡酰基奎宁酸和4,5二咖啡酰基奎宁酸的含量分别为0.18%、0.26%、0.25%、0.30%、4.52%、2.78%、4.36%。
1.2.2 细胞用羊耳菊提取物储备液的制备 称取羊耳菊提取物5 g,加入适量DMSO超声溶解后,加入PBS定容至25 mL,即浓度为200 mg/mL的提取物母液(DMSO最终浓度为20%),0.22 μm的微孔滤膜过滤后分装保存至-20 ℃,临用前用完全培养基稀释成所需浓度。
1.2.3 细胞培养
1.2.3.1 细胞复苏 紫外照射超净台15~30 min,棉花用75%乙醇喷湿后,擦拭超净工作台,从-80 ℃冰箱将冻存的Raw264.7细胞迅速取出,将冻存管置于37 ℃水浴锅中,在1 min内迅速解冻,不停震摇的同时直至细胞液体完全溶解,75%乙醇喷湿冻存管后,拿入超净台内,酒精灯烤瓶盖周围,将冻存管内的液体转入10 mL离心管,加入3 mL含10% FBS的DMEM,1 000 r/min离心5 min,弃上清液,加入5 mL完全培养基吹打均匀后转移至25 cm2培养瓶中,于37 ℃,5% CO2培养箱中培养。
1.2.3.2 细胞传代 待细胞贴壁生长至90%后,弃掉瓶中培养基,PBS清洗2遍,然后加入1 mL 0.25%胰蛋白酶消化液,消化3~5 min后,用1~2 mL含10% FBS 的DMEM终止反应,沿瓶壁将细胞轻轻吹打下来,转移至10 mL离心管中,1 000 r/min,5 min离心,按1 ∶6传代至新的培养瓶中,于37 ℃,5% CO2培养箱中培养36~48 h后,稳定传代几次后,进行接下来的实验。
1.2.3.3 细胞冻存 待细胞生长稳定,弃掉瓶中培养基,PBS缓冲液清洗两遍,然后加入1 mL 0.2%胰蛋白酶消化液,消化3~5 min后,用1~2 mL 含10% FBS的DMEM终止反应,沿瓶壁将细胞轻轻吹打下来,转移至10 mL离心管中,1 000 r/min,5 min离心,加入1 mL冻存液(FBS ∶DMSO=9 ∶1),梯度冻存细胞,即4 ℃冰箱放置30 min,-20 ℃放置2 h,转入-80 ℃长期冻存。
1.2.4 溶液配制
1.2.4.1 内标溶液的配制 精密称取葛根素5.7 mg,加甲醇定容至5 mL棕色容量瓶中,得1.14 g/mL的葛根素储备液,后续根据实验需要将其制备成实验所需浓度,存放至-20 ℃备用。
1.2.4.2 对照品溶液的配制 精密称取木犀草苷5.07 mg、绿原酸10.13 mg、新绿原酸11.16 mg、隐绿原酸10.28 mg、3,4-二咖啡酰基奎宁酸15.21 mg、3,5-二咖啡酰基奎宁酸15.33 mg, 4,5-二咖啡酰基奎宁酸15.17 mg,加甲醇分别定容至5 mL,分别得到浓度为1.01、2.01、2.22、2.05、3.02、3.03、3.01 mg/mL的标准储备液,保存在-20 ℃备用;临用前逐步将储备液稀释成混合标准溶液(木犀草苷、绿原酸、新绿原酸、隐绿原酸、3,4-二咖啡酰基奎宁酸、3,5-二咖啡酰基奎宁酸、3,4-二咖啡酰基奎宁酸浓度分别为61.76、124.18、124.24、124.41、491.25、249.08、496.60 ng/mL),再按倍数稀释至表1中的系列浓度的标准溶液。
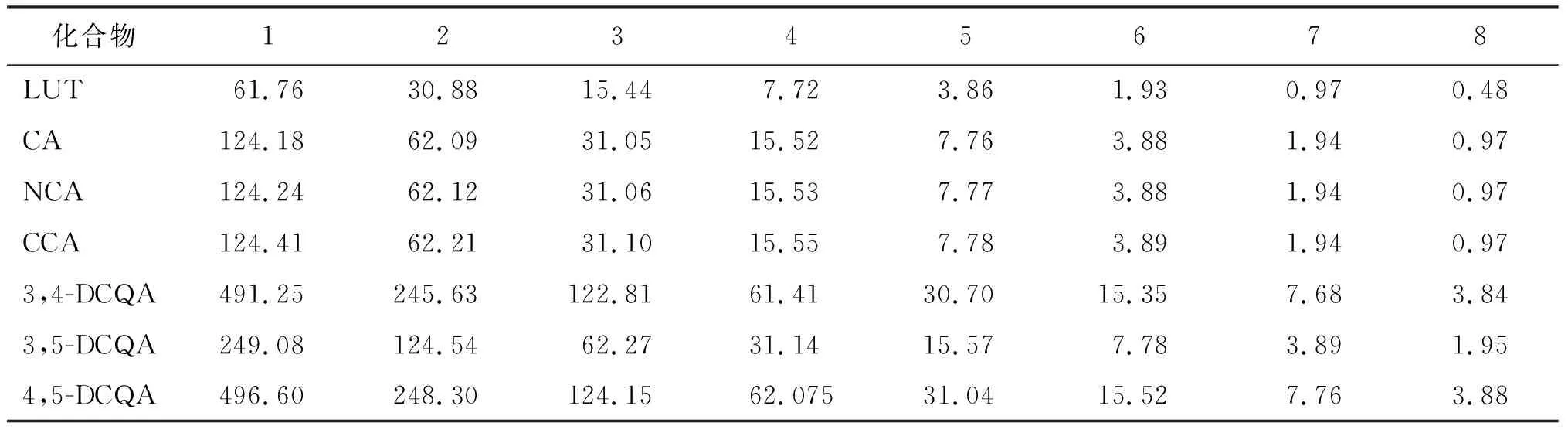
表1 木犀草苷等7个活性指标成分系列标准溶液浓度 ng/mL
1.2.4.3 PBS配制 将规格为2 L每袋的PBS,加入超纯水溶解后,补至2 L,分装在蜀牛瓶中后,高压蒸汽灭菌后4 ℃存储使用。
1.2.5 细胞样品处理方法 取800 μL经过3次冻融的细胞悬液,加50 μL葛根素溶液(15 ng/mL),短暂涡混后加160 μL 20%甲酸水溶液和800 μL甲醇,涡混5 min,超声10 min,在4 ℃条件下12 000 r/min离心10 min,上清液使用氮吹仪吹干,150 μL 50%甲醇复溶残渣部分,涡混5 min,超声10 min,在4 ℃条件下14 000 r/min离心10 min,上清液进样分析。
1.2.6 UPLC-MS/MS分析方法的建立
1.2.6.1 液相条件 色谱柱:Waters BEH C18(2.1 mm×50 mm,1.7 μm)柱;保护柱:Waters Van Guard BEH C18(2.1 mm× 5 mm,1.7 μm);流速为0.35 mL/min;柱温为40 ℃;流动相:0.2%甲酸水(A)-0.2%甲酸乙腈(B);进样体积为3 μL。梯度洗脱条件见表2。

表2 木犀草苷等7个活性指标成分的色谱条件
1.2.6.2 质谱条件 采用电喷雾离子源(ESI),毛细管电离电压为3 kV,离子源温度为150 ℃,喷雾气与反吹气是N2,去溶剂气流速为1 000 L/h,去溶剂气温度为600 ℃,扫描方式为多反应监测模式(MRM),质谱数据采集及处理软件为MassLynx V4.1工作站。木犀草苷等7种成分及内标用于定量分析的监测离子见表3。

表3 木犀草苷等7个活性指标成分及内标的质谱条件
1.2.7 分析方法的确证
1.2.7.1 专属性 取800 μL空白细胞悬液按“1.2.5 细胞样品处理方法”项下方法操作(不加内标),得空白图谱A;将一定浓度对照品溶液和葛根素内标溶液加入空白细胞悬液,按“1.2.5”方法操作获得对照品图谱B;取给药后的细胞悬液,按“1.2.5”方法操作得含药谱图C。
1.2.7.2 标准曲线的制备和LOD、LOQ 取800 μL空白细胞悬液,依次加入含有木犀草苷等7种成分的混合系列标准溶液50 μL,按“1.2.5细胞样品处理方法”项下操作。以待测成分与内标峰面积之比(A/Ai)为纵坐标Y,各成分质量浓度(C)为横坐标X进行线性回归。木犀草苷等7种成分的最低定量限(LOQ)定义为S/N=10,最低检测限(LOD)定义为S/N=3。
1.2.7.3 准确度和精密度 取800 μL空白细胞悬液,分别配制木犀草苷等7种成分的低、中、高三个浓度的QC样品(n=5),取50 μL加入后,其余均按“1.2.5”方法操作,低、中、高三个浓度同一天连续进样2次,连续测定3 d,分别算出日内精密度和日间精密度。
1.2.7.4 提取回收率与基质效应 分别配制A、B、C三种样品,取800 μL空白细胞悬液,配制木犀草苷等7种成分的空白细胞悬液中浓度的QC样品,平行进行5样本分析,取50 μL加入后,其余按“1.2.5”方法操作,此操作得到的样品为A;另取一份800 μL空白细胞悬液,不加混合对照品溶液和内标外,甲酸和甲醇按“1.2.5”项下操作,离心后,取上清液,上清液中加入与A样品中相对应浓度的对照品,每种浓度进行5样本分析,吹干后,150 μL 50%甲醇复溶,此样品为B;C样品中不加入空白细胞,取上述中相应浓度的对照品溶液加入,按“1.2.5”项下操作。结果计算时,7种成分的提取回收率为样品A峰面积与样品B峰面积之比,基质效应为样品B峰面积与样品C峰面积之比。
1.2.7.5 稳定性 取800 μL空白细胞悬液,分别配制木犀草苷7种成分低、中、高浓度的QC样品,取50 μL加入后,其余按“1.2.5”项下操作,分别观察样品在自动进样器中分别放置12 h,-20 ℃下冷藏24 h后的稳定性,以每一浓度5样本分析。
1.2.8 含药细胞悬液的制备 选取对数生长期的细胞进行计数,将细胞悬液的浓度调整至1×105个/孔,接种至96孔板,每孔加100 μL细胞悬液,混匀后放置于CO2培养箱培养24 h,弃掉培养基,加入PBS轻轻清洗2次,加入100 μL含600 μg/mL羊耳菊提取物药液的10% FBS的DMEM,设置6个复孔,放置于CO2培养箱培养10 h,弃药液,加入PBS轻轻清洗3次,收集细胞,然后细胞用2 mL预冷的PBS快速洗涤2次,然后向每个孔中加入2 mL超纯水,并将样品储存在-80 ℃,经过3次冷冻和解冻后,获得含药细胞悬液。
2 结果
2.1 专属性
在选定的色谱和质谱条件下,木犀草苷等7种成分在细胞悬液中的分离良好,且无杂质干扰。空白细胞悬液、空白细胞悬液加入混合对照品溶液及葛根素内标和给药后细胞悬液样品色谱见图1。

1: 新绿原酸; 2: 绿原酸; 3: 隐绿原酸; 4: 葛根素(内标); 5: 木犀草苷;6: 3,4-二咖啡酰基奎宁酸; 7: 3,5-二咖啡酰基奎宁酸; 8: 4,5-二咖啡酰基奎宁酸
2.2 线性范围与LOQ、LOD
细胞悬液中木犀草苷等7种成分在浓度范围内线性关系良好,相关系数(r2)均大于0.99,细胞悬液标准曲线方程及最低定量限(LOQ)、最低检测限(LOD)见表4。
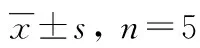
表4 木犀草苷等7种成分在细胞悬液中的线性回归方程
2.3 准确度和精密度
对低、中、高三个浓度的细胞悬液的日内精密度和日间精密度进行了考察,结果表明木犀草苷等7种成分日内精密度和日间精密度RSD均小于15%,低、中、高浓度的准确度范围为90.78%~113.99%,说明该方法准确、可靠、重现性好,测定结果见表5。
表5 木犀草苷等7种成分在细胞悬液中的准确度与精密度
2.4 提取回收率与基质效应
对木犀草苷等7种成分线性范围内低、中、高浓度的细胞悬液样品提取回收率进行了考察,结果表明在浓度范围内7种成分的回收率及基质效应符合生物样品分析方法要求。具体结果见表6。

表6 木犀草苷等7种成分在细胞悬液中的提取回收率与基质效应
2.5 稳定性
对木犀草苷等7种成分低、中、高浓度细胞悬液样品在进样器放置12 h,冷藏(-20 ℃)24 h的稳定性(n=5)进行了考察,结果表明7种成分在细胞悬液中经过上述过程后均稳定,具体结果见表7。
表7 木犀草苷等7种成分在细胞悬液中的稳定性
3 讨 论
3.1 细胞破碎方法的选择
细胞常用的破碎方法有物理裂解方法和化学裂解方法,其中化学裂解方法是使用裂解液裂解细胞,比如RIPA裂解液[10]、IP细胞裂解、NP-40裂解液、SDS裂解液等,由于此类型的细胞裂解液中大部分成分是盐类,使用后会在UPLC-MS/MS仪器管路中产生盐析,堵塞仪器,影响仪器使用寿命;物理裂解方法主要有超声裂解[11]和反复冻融[12-13]方式,超声裂解仪可以充分将细胞破裂,但该方法耗时,不适合大批量处理细胞,且实验前期进行过预实验,发现两种破碎方式进样测定后细胞中药物含量差别不大。因此,本实验选用简便,可批量处理的反复冻融方式进行破碎细胞。
3.2 样品蛋白沉淀方法选择
常见的样品蛋白沉淀方法有液-液萃取法、固相萃取[14]、蛋白沉淀等多种样品处理方法,课题组前期已建立了羊耳菊提取物中活性成分体内含量测定的超高效液相色谱法串联质谱的方法[15-17],考虑到细胞样品中药物浓度过低、基质不同等问题,进行了优化并验证,建立了快速精准的测定细胞内咖啡酸类含量的分析方法,又加之样品处理数量大,选择萃取方法操作复杂,耗时耗力,故在此基础上选择了方法简便的甲醇沉淀蛋白。
3.3 细胞中咖啡酸类成分提取方法选择
羊耳菊抗炎有效部位中待测成分主要为咖啡酸类等化合物,在进行细胞处理时,需要用甲酸酸化样品,预实验中考察了1%、5%、10%、20%浓度的甲酸对细胞样品中咖啡酸类提取,发现20%浓度甲酸可以明显增加样品中绿原酸等7个活性指标成分的含量,提高检测灵敏度。
综上,本文建立了快速、准确的同时测定Raw264.7细胞中羊耳菊提取物所含的7个指标成分(LUT、CA、NCA、CCA、3,4-DCQA、3,5-DCQA、4,5-DCQA)含量的UPLC-MS/MS分析方法。同时,本次实验中,对细胞样品的专属性、线性、准确度、精密度、提取回收率、基质效应和稳定性进行考察,发现该分析方法符合细胞样品的测定,为开展羊耳菊提取物的细胞药代动力学研究奠定基础。
猜你喜欢
山东第一医科大学(山东省医学科学院)学报(2020年10期)2020-11-20 07:56:54
分析测试学报(2020年4期)2020-05-09 08:13:14
发酵科技通讯(2018年2期)2018-07-06 11:38:46
天然产物研究与开发(2018年2期)2018-04-04 02:01:12
西江月(2017年4期)2017-11-22 07:24:09
长春中医药大学学报(2017年1期)2017-04-16 05:56:45
小雪花·成长指南(2016年10期)2016-11-01 06:02:27
知音·下半月(2016年5期)2016-05-27 13:01:31
中国野生植物资源(2014年1期)2014-03-29 05:45:02
中成药(2014年10期)2014-02-28 22:29:40
