
Barth综合征两兄弟及其TAZ基因新突变*
2022-11-27 06:26宋金青张立红杨艳玲
罕少疾病杂志 2022年11期
许 蓓 宋金青 刘 怡 金 颖 张 尧 张立红 杨艳玲,*
1.河北省保定市第一中心医院普儿科 (河北 保定 071000)
2.北京大学第一医院儿科 (北京 100034)
Barth综合征(OMIM 302060)是一种罕见的X连锁遗传代谢病,为TAZ基因致病变异导致的有机酸代谢病,1983年由Barth首次报道[1]。本病通常在婴儿期起病,以心肌病、骨骼肌病变、中性粒细胞减少、发育落后为主要表现,生化异常特征为3-甲基戊烯二酸尿症[2]。由佛罗里达和波士顿儿童医院发起的Barth综合征基金会注册登记的Barth综合征存活者约200人,估计活婴中的患病率为1/300000~1/400000[3]。我国上海儿童医学中心对2012~2015年就诊的180名儿童心肌病患者进行了二代测序基因分析,其中TAZ突变率为3.5 %[4]。Barth综合征临床表现缺乏特异性,极易漏诊、误诊,是男性胎儿流产、死胎和婴幼儿死亡的原因之一[5]。本文通过对TAZ基因新突变的导致Barth综合征两兄弟的研究,探讨Barth综合征急性代谢危象的诱发因素、临床表现、诊断、治疗、遗传咨询及预防。
1 资料与方法
2例患儿为同胞兄弟,父母体健,非近亲婚配,既往无不良产史及家族性疾病史。病例1:男,10个月时夭折。患儿为G1P1,足月顺产出生,出生体重3Kg,母乳及配方奶粉混合喂养,迁延性腹泻,稀便7~8次/日。智力发育正常,运动发育落后,4~5个月时能翻身,10个月时不能独坐,不能爬,生长迟缓,未出牙,体重增加不良。10个月时注射疫苗,次日起嗜睡,纳差,拒奶,3天后手脚浮肿,6天后患儿哭闹不止,气促,呼吸困难,心跳加快。当地医院超声心动图检查发现扩张型心肌病、心力衰竭,化验发现低血糖,抢救无效死亡,疑诊“心肌病、多脏器功能衰竭”,曾进行糖原累积症相关基因筛查未见异常。病例2:男,病例1的胞弟,G2P2,足月顺产出生,出生体重2.68 Kg。5个月时因“体重不增、生长迟缓”来院就诊。患儿以母乳及配方奶粉混合喂养,初始喂养困难,稀便7~8次/日,3个月后体重不增,5个月时体重仅5.7 kg,改用免乳糖配方奶粉、加米糊后腹泻好转。运动发育落后,2个半月可竖头,可逗笑,可追光、追物,肢体软,睡眠中易惊,无惊厥,5个月时不会翻身,智力正常。3个月时接种百白破疫苗,24小时后发热,肢体无力。
2 结 果
2.1 常规化验检查血液中性粒细胞数值显著降低(0.25×109/L,参考范围2.3~7.7×109/L),白细胞总数正常(4.86×109/L),红细胞总数正常(4.45×1012/L),血红蛋白正常(123 g/L),血小板332×109/L,淋巴细胞数值3.66×109/L。血清肌酸激酶正常(88IU/L,参考范围25~195 IU/L) ,肌酸激酶同工酶稍高(7.7ng/mL,参考范围<5ng/mL),肌钙蛋白增高(0.34ng/mL,参考范围0~0.03ng/mL)。N端脑钠肽前体明显升高(2182pg/mL,参考范围<450pg/mL)。
2.2 放射检查胸部正位片:心影增大,心胸比0.58。
2.3 超声检查超声心动图:左心室心肌均匀肥厚,左室心尖肌小梁显著增多,左室收缩功能正常范围,见图1。
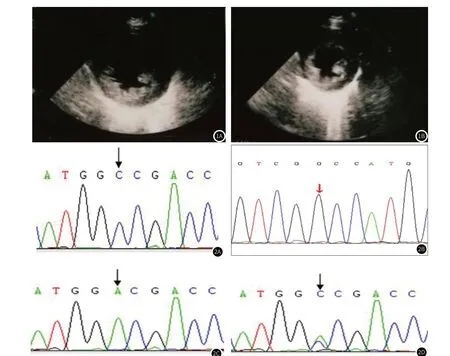
图1 图1A~图1B:病例2,超声心动图,各房室内径大致正常,左室心尖段各壁可见肌小梁层,最厚处于下壁,非致密层(NC)/致密层(C)5.8/4.9mm。左心室心肌均匀肥厚,回声均匀减弱,左室壁运动幅度正常。左室心尖肌小梁显著增多,左室收缩功能正常范围。图2 Barth综合征两兄弟及其父母TAZ基因部分测序图,图2A:弟弟TAZ基因c.221A>C;图2B:哥哥 TAZ基因c.221A>C;图2C:患者父亲 TAZ基因无c.221A>C;图2D:患者母亲 TAZ基因c. 221A>C。
2.4 血、尿代谢筛查血液氨基酸及酯酰肉碱谱未见异常。尿有机酸分析显示3-甲基戊烯二酸升高(7.02mg/g肌酐,参考范围0~4.2mg/g肌酐),提示3-甲基戊烯二酸尿症。
2.5 基因分析经家属知情同意,抽取病例2及父母静脉血各3毫升,取病例1去世之前留存的血样,提取基因组DNA,应用高通量全外显子测序技术进行基因检测。发现病例1、病例2两兄弟染色体Xq28区域TAZ变异c.221A>C,引起TAZ蛋白质第74位氨基酸错义突变p.D74A(NM_000116)。运用Sanger技术进行家系验证,TAZ变异c.221A>C来源于患儿母亲(图2),符合X连锁遗传病。TAZ c.221A>C在正常人数据库(DYDF,dbSNP,千人基因组,千人南方,千人北方,EXAC)中未收录,为尚未报道的新变异,导致蛋白编码的异常翻译,蛋白结构预测为可能致病性变异。结合两兄弟临床表现、3-甲基戊烯二酸尿症及心肌病表现,确诊为Barth综合征。
2.6 治疗与随访病例2确诊后开始治疗:(1)营养支持,低乳糖饮食,补充益生菌、中链脂肪酸及维生素A、D等维生素。(2)营养心肌,改善心功能,口服辅酶Q10、酒石酸美托洛尔、地高辛和利尿剂等。(3)口服左卡尼汀,促进有机酸排泄。(4)避免疲劳,如有感染积极控制,心功能正常、心肌恢复正常后补种计划内疫苗。
随访4年1个月:患儿运动能力改善,1岁可扶站、扶走,超声心动图检查左室收缩功能基本正常。1岁7个月独走,易疲劳。2岁3个月会说话,会跑。现在患儿4岁6个月,精神、食欲良好,体力稍差,智力正常,在幼儿园中班就读,适应能力良好。
2.7 遗传咨询及父母再生育指导母亲健康,超声心动图正常。第三胎妊娠中期接受产前诊断,羊水细胞TAZ基因未检出c.221A>C变异,胎儿为女婴,足月顺产出生,现在2岁,发育良好。
3 讨 论
Barth综合征是一种多系统疾病,以心肌病、骨骼肌病变、发育迟缓、生长迟缓、中性粒细胞减少及3-甲基戊烯二酸尿症为主要特征。随着临床识别能力的提高,发现Barth综合征更多复杂的表型。有的患儿稳定期无明显的粒细胞减少及3-甲基戊烯二酸尿症,极少数甚至不伴有心肌病[6],但是,在感染、疲劳、预防接种等应激状态下出现代谢危象及心肌病,为疾病的防治带来了很大挑战。2010年Steward CG首次报道了Barth综合征导致的婴儿猝死和胎儿死亡[5],迄今我国报道Barth患者不足10例[7-8]。
编码Tafazzin的TAZ基因突变是导致Barth综合征主要遗传缺陷,TAZ基因位于Xq28,无义突变或缺失突变均可造成心磷脂重塑缺陷,导致心肌病。心磷脂位于线粒体内膜,对稳定线粒体呼吸链功能起重要作用[9]。TAZ基因序列在进化中高度保守,共包含11个外显子,目前共发现180多种不同的突变,大部分为错义突变和小的插入或缺失突变[3]。本文两兄弟均为TAZ c.221A>C,为新的错义突变,来源于其母亲,符合X连锁遗传病规律。然而,本病基因型和表现型的关系不明,同一家庭内男性的表现型也有差别。Ronvelia D[10]等曾报道一个家系,先证者(51岁男性)和其妹妹的儿子TAZ外显子7存在相同的突变 c.583G>T,先证者幼时仅表现为肌无力,45岁时才发现左心室致密化不全,而其妹妹的儿子喂养困难,肌张力降低,伴酸中毒和低血糖,3个月后体重不增,嗜睡,呼吸困难和心力衰竭,11个月时进行了心脏移植,随访到3岁,超声心动图显示患儿心脏大小和心功能正常,生长发育逐渐进步。
心肌病是Barth综合征最主要的临床表型,多于婴幼儿期发病。常见扩张型心肌病伴左室肌小梁增粗、左心室致密化不全,少数病例表现为心肌肥厚,也有一个家系中同时存在肥厚型和扩张型心肌病[6,11]。左心室壁可随年龄发生“重塑”,在相对扩张和肥厚之间变迁,一个左心室致密化不全的患者也可演变成“非扩张型”表型[12]。本研究中两同胞兄弟心脏改变有所不同,胞兄心脏扩大,而其弟心肌肥厚。Barth综合征患者可随时出现室性心律失常和猝死,与心肌病的程度无关[13]。伴有心衰的患者,应用血管紧张素转换酶抑制剂、酒石酸美托洛尔、地高辛和利尿剂药物治疗有效[6],也有一些患者接受了心脏移植,预后较好[3]。
Barth综合征患者由于营养不良、腹泻、心肌病、口腔溃疡和反复感染,常有显著的生长迟缓。本研究中2例患儿喂养困难,发育落后,体重不增,迁延性腹泻,弟弟体力更差,符合Barth综合征特征。由于中性粒细胞降低,免疫力下降,Barth综合征患者易患感染性疾病,约半数患者死于感染,并不是死于心脏合并症[14]。本研究中哥哥病情进展迅速,10个月时夭折,病因不明,经冻存血样基因分析确诊为Barth综合征。其胞弟早期检查,存在严重的中性粒细胞缺乏,伴心肌肥厚及发育落后,符合Barth综合征。大多数Barth综合征患者尿3-甲基戊烯二酸升高,也被称为3-甲基戊烯二酸尿症II型。然而,也有一些TAZ突变的Barth综合征患者尿液3-甲基戊烯二酸正常或者呈波动性增高[15],极易漏诊。
Barth综合征容易被漏诊、延误诊断,当患儿合并感染伴心肌病时,容易被诊断为病毒性心肌炎,患中性粒细胞减少时往往被认为病毒感染导致的骨髓造血抑制。病情相对稳定后,又被认为是病毒性心肌炎恢复期[14,16]。所以,对于心肌病患者应注意Barth综合征,尤其是男性婴幼儿心肌病患者。尿有机酸分析、TAZ基因分析是确诊Barth综合征的关键,多数患者血液游离肉碱降低,羟异戊酰肉碱增高,有助于疾病筛查及诊断,全外显子捕获和高通量测序技术准确性高,速度更快,有助于提高Barth综合征的基因诊断率。
预防接种是儿童保健的重要组成部分,可有效预防多种严重感染性疾病。但是,对于接种疫苗前尚未发病的某些先天缺陷患儿,可能诱发发病,甚至猝死[17-18]。Barth综合征患者常有中性粒细胞减少,免疫功能缺陷,在发热、饮食不当、疲劳、药物、饥饿等应激状态下可急性起病[19-20]。对于这类患儿应慎用活疫苗,待病情稳定后接种灭活疫苗,接种疫苗后应检测血清抗体水平[21-22]。本文两兄弟均为疫苗接种后起病,其兄病情迅速恶化死亡。其弟接种疫苗后也出现了发热、乏力、肢体软等症状。
Barth综合征是一种潜在的致死性心肌病,大部分患儿死于心力衰竭或者严重感染。据报道,70%的Barth综合征患者在未确诊前死亡,如果能早期诊断并治疗,将使病死率降到10%[15]。通过积极治疗,部分患者可长期存活,报道最年长的患者已经50多岁[23]。Barth综合征为X连锁遗传病,女孩多为表型正常的携带者,TAZ突变男性为患者。对于Barth综合征家族,应给予遗传学指导,母亲再孕时争取产前诊断,通过胎盘绒毛或羊水细胞TAZ基因分析进行胎儿诊断[2,4-5]。
猜你喜欢
分子催化(2022年1期)2022-11-02
化学工业与工程(2022年1期)2022-03-29
中成药(2019年12期)2020-01-04
中国临床医学影像杂志(2019年1期)2019-04-25
天然产物研究与开发(2018年10期)2018-11-06
天然产物研究与开发(2018年10期)2018-11-06
中国塑料(2016年2期)2016-06-15
中国医学科学院学报(2015年5期)2015-03-01
中国当代医药(2015年31期)2015-03-01
中国医药科学(2015年15期)2015-02-27
