
环丙沙星和铜复合污染下河流底质微生物群落与抗生素抗性基因的交互关系
2022-11-25 13:24:50王琳琼范晨阳
河海大学学报(自然科学版) 2022年6期
李 轶,胡 童,王琳琼,范晨阳
(1.河海大学环境学院,江苏 南京 210098;2.河海大学浅水湖泊综合治理与资源开发教育部重点实验室,江苏 南京 210098;3.河海大学海洋学院,江苏 南京 210098)
抗生素具有高效、强力、广谱的杀菌特性,在医疗、畜牧、养殖等领域均有广泛的应用。根据中华人民共和国国家卫生健康委员会统计,2019年我国抗生素使用量达到16.1万t。人和畜禽对抗生素的利用率有限,摄入的抗生素约有40%会排出体外,这些含有抗生素的污水部分被直接排入水环境中[1-2]。尽管环境中微生物可对抗生素等有机物进行转化和降解,但由于其降解率较低[3],导致其在环境中累积。大量研究表明,抗生素的过量使用是抗生素抗性基因 (antibiotic resistance genes,ARGs) 在环境中出现和广泛传播最主要和直接的原因[4]。通过可移动遗传元件 (mobile genetic elements,MGEs) ,ARGs从基因组转移到噬菌体或质粒,从而促进了水环境中耐药细菌 (antibiotic resistance bacteria,ARB) 和ARGs的增殖和传播[5]。
河流底质为微生物提供了优良的栖息场所,是水环境中微生物生命活动最活跃的区域,同时微生物也是河流中物质循环的主要参与者,决定着河流生态系统的稳定性[6]。ARGs作为一种基因层面的污染物,其传播和扩散与微生物的生命活动存在联系。微生物群落与ARGs之间有着复杂的相互作用机制,微生物群落组成的变化会影响ARGs宿主或敏感菌的丰度变化[7-8]。反之,ARGs的大量排入会对环境中土著微生物群落造成影响,获得ARGs的菌种可能被赋予更强的生存优势,再加上外源污染的干扰,最终导致微生物群落组成的变化[9-10]。此外,在一定条件下,ARGs会在不同种属的细菌间传播,一旦人类致病菌获得ARGs,就可能会对抗生素产生耐药性,导致抗生素治疗的失效,威胁人类健康[11]。
河流底质作为众多污染物的“汇”,抗生素、重金属等污染物被频繁检出。胡冠九等[12]对江苏省某市河流中抗生素浓度进行检测分析,结果表明磺胺类含量相对最高 (51.9%),其次是四环素类 (29.9%) 和喹诺酮类 (14.5%),其中磺胺类药物最高质量浓度可达 52.7 ng/L。封梦娟等[13]对长江南京段水源水中14种抗生素进行测定,平均质量浓度为 0.14~49.91 ng/L,其中克林霉素作为首要污染抗生素,其质量浓度高达 739.44 ng/L。王龙飞等[14]研究表明,在河流底泥和附着生物膜中磺胺类抗生素质量浓度分别为NF (未检出)~47.3 ng/g和NF~3 759.1 ng/g。另外,重金属作为难降解污染物之一,在河流底质中浓度较高。肖艳春等[15]对河流底泥中重金属含量检测分析发现,在污水处理厂排污口的点位,Ni (223.81 mg/kg) 和Cd (20.17 mg/kg) 质量分数最高,而Zn和Cu呈现空间差异性,从河流上游至下游其质量分数逐渐增加,最高值分别可达284.55 mg/kg和95.47 mg/kg。河流底质环境中污染物的赋存直接影响底质微生物群落的多样性、结构及功能,进而影响ARGs的分布及传播。
抗生素在河流环境中作为主要的相关性因素可直接促进相关ARGs的增殖与传播,同时还可能与其他类抗性基因存在联系[16]。尽管大多数环境中抗生素的残留处在一个较低的水平,但仍会对ARGs有选择作用[17]。Yang等[18]对洞庭湖中的抗生素和ARGs含量进行了调查,8种抗生素的总质量分数(磺胺类4种和四环素类4种)在60.02~321.04 μg/kg之间,同时在底质样品中sul1、sul2、tetA、tetC和tetM等抗性基因均被检出,且ARGs、残留抗生素和底质特征之间存在显著的相关性。此外,河流底质中重金属的赋存同样会对ARGs施加选择压力,从而提高环境中ARGs的丰度[19-20]。有研究发现,环境中的重金属浓度与ARGs丰度呈正相关关系,表明ARGs丰度的增加也可能与重金属污染有关[21-22]。然而,目前关于抗生素和重金属复合污染条件下河流底质微生物群落特征及其与ARGs间的交互研究较少,两者间的相互作用关系仍有待进一步研究。
本文选择环丙沙星(ciprofloxacin,CFC)和铜(copper,Cu)为代表性抗生素和重金属,采集江苏省南京市秦淮河江宁段污水处理厂上游点位的底质和水体样品进行微宇宙试验。通过设计室内驯化试验,利用高通量测序和荧光定量PCR等生物信息学技术,检测分析微生物群落组成和ARGs丰度,通过部分冗余性分析、路径分析、网络分析等方法,分析微生物群落和ARGs对复合污染的响应,考察微生物群落特征对环境因子的响应及其与ARGs的交互关系,探究微生物群落及污染物对ARGs传播扩散的作用机制,从而为河流中ARGs的污染防控提供理论依据。
1 材料与方法
1.1 样品采集与处理
河流底泥样品于2021年9月8日取自于江苏省南京市秦淮河江宁段污水处理厂上游河流,采用抓斗式采泥器采集水泥界面向下0~20 cm处的底泥。将采集到的沉积物装入聚乙烯塑料袋中,避光封口后带回实验室,去除石块、水生动物和植物残体后混合均匀,随后转移到4 ℃冰箱内保存直至驯化试验待用。
1.2 试剂与器材
试剂:环丙沙星(生物级)、CuSO4·5H2O、Na2HPO4·2H2O、MgSO4·7H2O、NaHCO3、Na2CO3、NaNO3、Na2SO4、(NH4)2·SO4、KH2PO4、NaCl、KCl和NH4Cl均为分析纯,购于国药集团化学试剂有限公司;FastDNA Spin Kit for Soil(MP bio土壤基因组DNA提取试剂盒)、SYBR Green qPCR Mix(2×实时定量PCR预混液)、Platinum Taq DNA聚合酶、50×TAE缓冲液、6×DNA Loading Buffer、DL2000 DNA Marker和10×Ex Taq Buffer购于生工生物工程(上海)股份有限公司。
器材:无菌超净台、高速离心机(德国Eppendorf)、移液枪(1 μL、2 μL、10 μL、100 μL和1 000 μL)、电泳仪(BIO-RAD,美国)、凝胶成像仪(Tanon-3500,上海天能)、实时荧光定量PCR仪(ABI7500,美国Applied Biosystems)、漩涡振荡仪、水浴锅(HH-4,常州普天仪器制造有限公司)和高压灭菌锅等。耗材有2 mL离心管、不同规格的枪头、96孔PCR板、八连排PCR管等。所有耗材均经过高压灭菌锅灭菌处理后使用。
1.3 试验方法
1.3.1 试验设计
无机营养盐的配置:称取K2HPO4(3.80 g)、KH2PO4(1.00 g)、NaCl (1.00 g)、MgSO4(0.20 g)和NH4Cl (0.10 g)溶于1 L蒸馏水中,并用玻璃棒搅拌透明后,4 ℃保存待用。
驯化试验设置:准备15个500 mL的锥形瓶作为底质驯化培养的容器,共5个试验组,每组包括3个平行试验,且分别在第0 d、5 d、15 d、30 d采取底质样品。在每个锥形瓶中加入175 g已处理好的底质样品。以蒸馏水为溶剂分别配置10 mg/kg的CFC储备液和1 000 mg/kg的硫酸铜储备液,根据表1的最终质量分数设定值,量取3.5 mL CFC储备液至CFC1和CC1组的6个锥形瓶中,量取8 mL CFC储备液至CFC2和CC2组的6个锥形瓶中,量取16 mL硫酸铜储备液至Cu、CC1和CC2组的9个锥形瓶中,最后用无机营养盐将所有锥形瓶的净重补至200 g,搅拌均匀并贴上标签,用封口膜封住瓶口,在25℃黑暗环境中驯化培养。培养底质选择秦淮河上游点位作为驯化基质,质量分数设定参考秦淮河江宁区段CFC、Cu质量分数的最高值和最小抑菌浓度值(MIC),见表1。
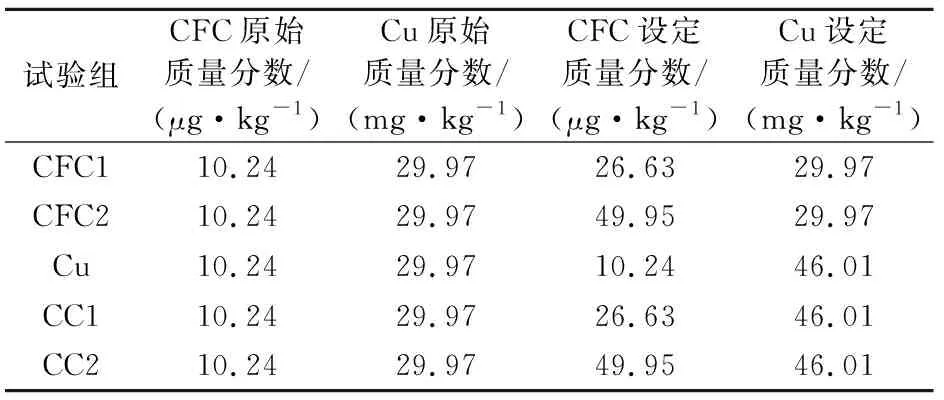
表1 室内驯化试验CFC和Cu质量分数设定
1.3.2 微生物测序及目标基因定量
底质样品DNA用FAST DNA®SPIN Kit for Soil试剂盒按操作说明提取。对样品进行DNA提取和质量检测后,预留荧光定量PCR所需的DNA样品(约10 μL),将剩余的DNA样品送至上海凌恩生物科技有限公司(Biozeren,上海)进行16S rRNA基因测序。待测序原始数据返回后,首先对原始数据序列进行优化处理。为了保证序列及分析结果的质量,通过QIIME(version1.17) 提取原始数据中的有效序列,并对数据进行除杂和过滤处理。按相似性大于97%的标准,将得到的优化序列进行OTU划分[23]。在区分样本后,进行OTU聚类分析和物种分类信息学分析。根据OTU聚类分析结果,计算Chao 1、Shannon、Simpson等微生物多样性指数;基于物种分类学信息,可以在各个分类水平上统计微生物群落组成和物种丰度。
室内驯化试验选取9种目标基因,分别为:16S rRNA、整合子intI1、转座子IS26、四环素类抗性基因tetA和tetT、氟喹诺酮类抗性基因QnrA和QnrC、磺胺类抗性基因sul1、大环内酯类抗性基因ermB,目标基因对应的正向引物、负向引物、定性和定量的退火温度如表2所示。目标基因PCR预试验选用30 μL混合的扩增体系,包括:15 μL qPCR Mix,2 μL Mg2+(25 mmol),两端引物各0.5 μL,灭菌超纯水10 μL和DNA模板2 μL。反应程序为95℃ 3 min加30个循环(94℃ 30 s;DNA退火温度30 s;72℃ 30 s)。反应后的PCR产物立即进行凝胶电泳检测。

表2 定量PCR方法进行分子检测的引物信息
目标基因的定量检测使用实时荧光PCR仪(Bio-Rad CFX96 Cycler Q5 thermocycler,USA),扩增采用20 μL体系,具体包括:10 μL 2 × SYBR green Premix Ⅱ (TaKaRa),两端引物各0.4 μL,灭菌超纯水8.7 μL和DNA模板0.5 μL。试验中的阴性对照组为反应体系中加入2 μL的ddH2O。荧光定量PCR采用标准曲线法,首先将已知浓度的标准品稀释10倍、102倍、103倍、104倍和105倍,将稀释后的基因标准品溶液作为模板进行PCR检测,得到目标基因的标准曲线。横坐标为基因标准品拷贝数,纵坐标为PCR仪检测得到的Ct值。在检测环境样品时,在同一个点位取3次DNA样品作为模板与标准样品梯度溶液一起检测。在相同试验检测条件下,得到环境样品目标基因的Ct值,代入已知浓度的标准品标准曲线即可计算得到样品中目标基因的拷贝数。目标基因的标准曲线方程、扩增效率和相关系数r2如表3所示。

表3 目标基因PCR定量标准曲线
1.4 数据处理
使用Office excel 2016软件进行数据处理和绘制图表,运用SPSS 17.0软件对样品进行Kruskal-Wallis H检验和Pearson相关性分析。当p<0.05时,认为结果具有显著性。组间菌种差异(LEfSe)分析在线上分析网站https://huttenhower.sph.harvard.edu/galaxy/进行。共现网络分析首先通过R语言psych包生成目标相关性矩阵,再通过Gephi0.9.2软件生成点、边文件,进而对点、边文件进行注释,最后在该软件导入点、边文件,调整相关参数,完成可视化。部分冗余性分析通过R语言计算。
2 结果与讨论
2.1 ARGs和微生物群落对复合污染的响应
2.1.1 CFC和Cu复合污染对ARGs丰度的影响
5个试验组目标ARGs在各取样时间点的相对丰度如图1所示。在所有样品中,总ARGs相对丰度为8.19×10-3~ 1.22×10-1copies/16S rRNA copy,最高相对丰度出现在CFC2试验组。从各试验组来看,CFC单一污染试验组(CFC1组和CFC2组)的总ARGs相对丰度随时间变化呈先上升(前15 d)后下降的趋势。在复合污染试验组(CC1组和CC2组)中,总ARGs相对丰度在第5天左右达到峰值,随后下降(p<0.05)。特别地,CFC单一污染试验组(CFC1组和CFC2组)和Cu单一污染试验组(Cu组)在总ARGs峰值上有显著的差异性(p<0.05)。ARGs丰度在CFC1组和CFC2组中的峰值最高,是Cu组的3倍。这可能是因为Cu的生物毒性造成底质微生物群落多样性和结构发生变化或致使特定种群消失,导致抗性基因的潜在宿主减少,从而对参与ARGs传播的细菌产生抑制作用[24-25]。
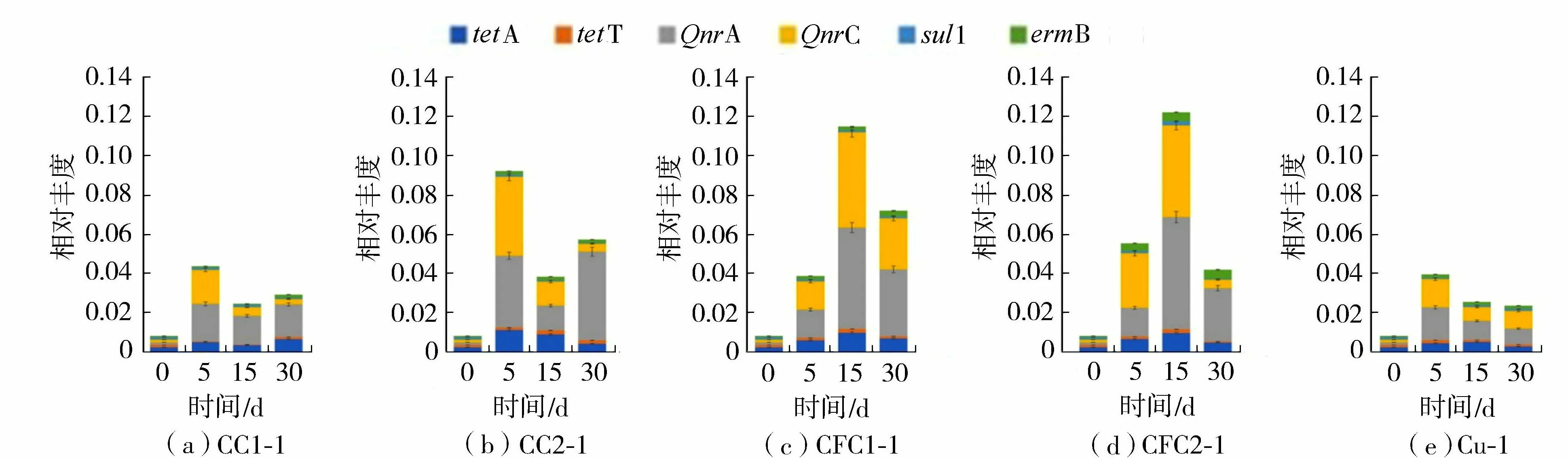
图1 各试验组ARGs相对丰度随时间的变化Fig.1 Changes of relative ARGs abundance in each experimental group with time
对比两个复合污染组(CC1组和CC2组),在浓度CFC为MIC值的CC2组中ARGs丰度更高,且每个采样时间点的ARGs丰度均高于CC1组。由此推断,CFC的MIC对ARGs增殖和传播具有一定的促进作用,且促进程度更大,表明环境中的MIC值对抗生素诱导ARGs具有重要的作用[26]。以上结果可解释为抗生素在一定条件下可被微生物转化或降解,当抗生素作为外界环境选择压力使得少数耐药菌获得更多生存资源而快速繁殖,从而加速了ARGs的增殖[3-17]。有研究表明,抗性基因丰度与抗生素及砷、铜等重金属污染浓度呈正相关关系,说明抗生素与重金属复合污染促进了环境中抗性基因的分布水平[27-28]。综上,通过非参数检验,表明5个试验组总ARGs相对丰度的时间分布呈显著差异,也说明不同的污染组在一定的时间段内影响ARGs的增殖和传播。
2.1.2 CFC和Cu复合污染对微生物群落结构及功能的影响
基于物种分类学分析,在门水平上对高通量测序结果进行归类,获得15个底质样品的微生物群落组成及相对丰度,结果如图2所示。共有15个丰度占比超过0.5%的菌门,其中超过5%的菌门有变形菌门(Proteobacteria)、酸杆菌门(Acidobacteria)、厚壁菌门(Firmicutes)、拟杆菌门(Bacteroidetes)和绿弯菌门(Chloroflexi),均为本试验样品中的优势菌门。变形菌门是所有样品中占据绝对优势的菌门,结合各试验组微生物群落的组成变化来看,变形菌门的丰度占比在添加Cu的试验组中随时间降低,在未添加Cu的试验组中没有发现相似的规律,说明较高的Cu浓度对变形菌门的丰度具有抑制作用。相反,厚壁菌门则在添加Cu的试验组中丰度占比随时间显著上升,这可能与厚壁菌门细菌的耐污能力有关,也使其成为抗生素和重金属复合污染环境下ARGs的潜在宿主[29]。酸杆菌门、拟杆菌门和绿弯菌门在各试验组中没有显著的时间变化规律。Tuo等[30]研究表明,环丙沙星对土壤优势微生物的影响较小,不同浓度环丙沙星污染组中门水平微生物丰度有显著差异,各处理优势门水平微生物及其丰度是放线菌门(Actinobacteria)、变形菌门、厚壁菌门和奇古菌门(Thaumarchaeota),该结果与本研究结果类似。
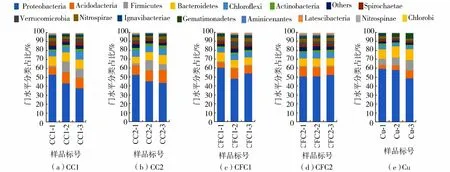
图2 各试验组样品在门水平上的微生物群落组成Fig.2 Microbial community composition of each sediment sample at the phylum level
通过对各样品的OTU组成进行PCoA分析(图3),进一步辨别各试验组样品微生物群落组成之间的差异性。图3表明各样品的整体分布与污染类型显著相关,CFC和Cu复合污染的两个试验组CC1和CC2的6个点聚集在图上方,CFC单一污染试验组的点都在图右侧,Cu单一污染组除1个点外均聚集在右下角,表明不同的污染类型是造成微生物群落组成差异性的重要原因。已有研究发现,复合污染和单一污染对微生物群落组成的影响不同,多数抗生素和重金属复合污染对微生物群落多样性和结构产生显著的影响,呈现微生物群落组成的差异性[31]。侯俊等[32]探究了人工湿地系统中有机污染物对微生物群落的影响,结果表明污染物可直接改变微生物群落的丰度和多样性,相反微生物通过降解有机污染物,从而提高了微生物群落的耐受性。孙依欣[25]认为,镉与环丙沙星的单一污染和复合污染试验组中微生物群落结构呈现明显的差异,单一环丙沙星处理组使得微生物群落多样性增加,而复合污染处理组中微生物群落多样性呈现下降趋势。
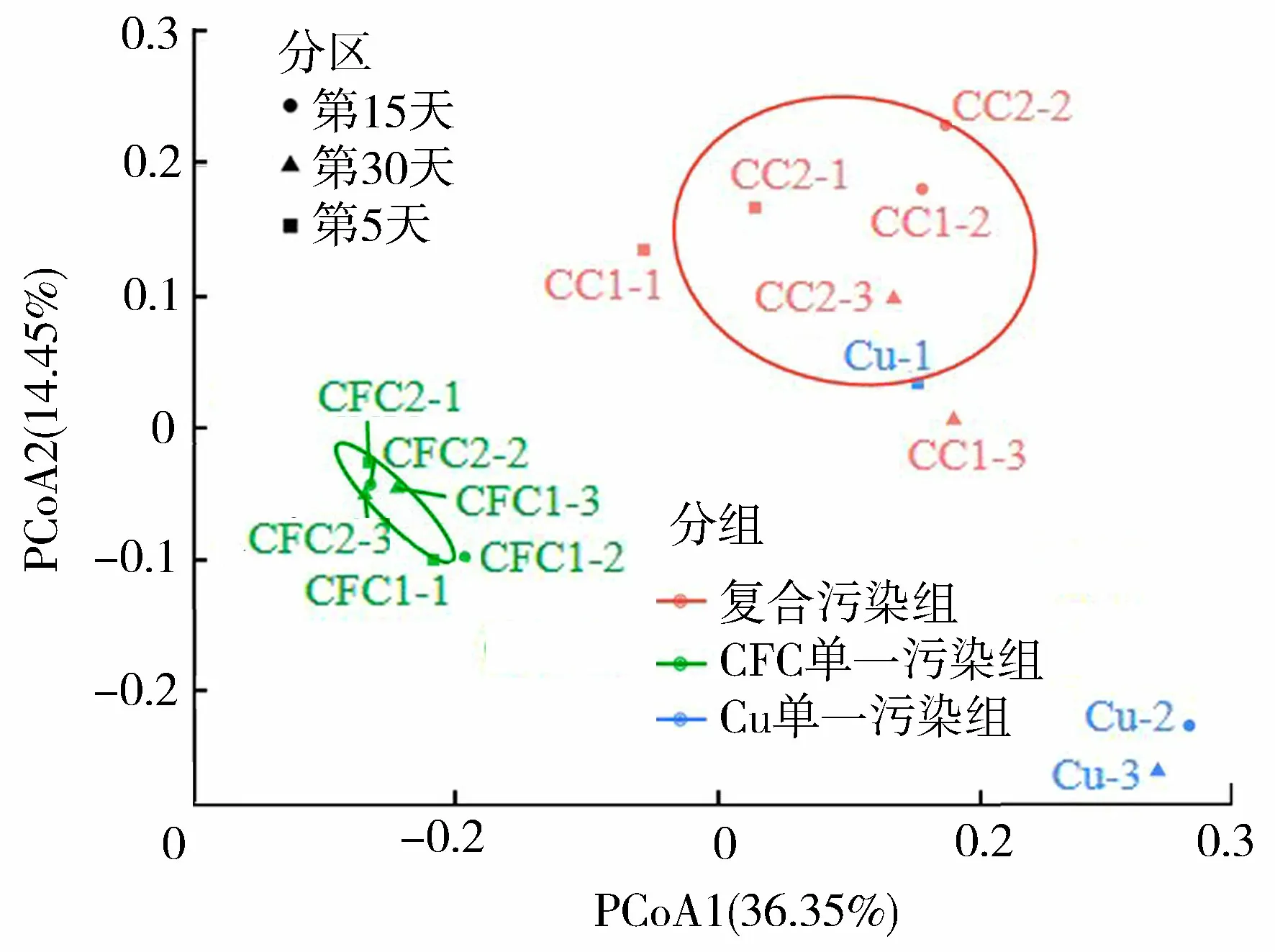
图3 各试验组微生物群落PCoA分析Fig.3 PCoA analysis of microbial community in each experimental sample sediment
另外,将5个试验组的高通量测序数据与KEGG数据库进行对比,得到各样品组的一级功能预测结果,如表4所示(不包括未分类序列)。由表4可见,新陈代谢、遗传信息加工和环境信息处理是5个试验组中微生物群落的主要功能,相对丰度占比分别为48.52%~49.46%、16.56%~17.21%和12.02%~13.24%。各试验组一级功能预测结果无显著差异。如图4所示,二级功能预测结果表明,氨基酸代谢和其他次生代谢产物的合成在CFC单一污染组中的丰度明显高于复合污染组;复合污染组和Cu单一污染组功能的丰度组成相似,无显著差异,表明当CFC浓度不超过MIC值时,污染物CFC对底质微生物群落的氨基酸代谢和其他次生代谢的代谢产物合成均具有促进作用。
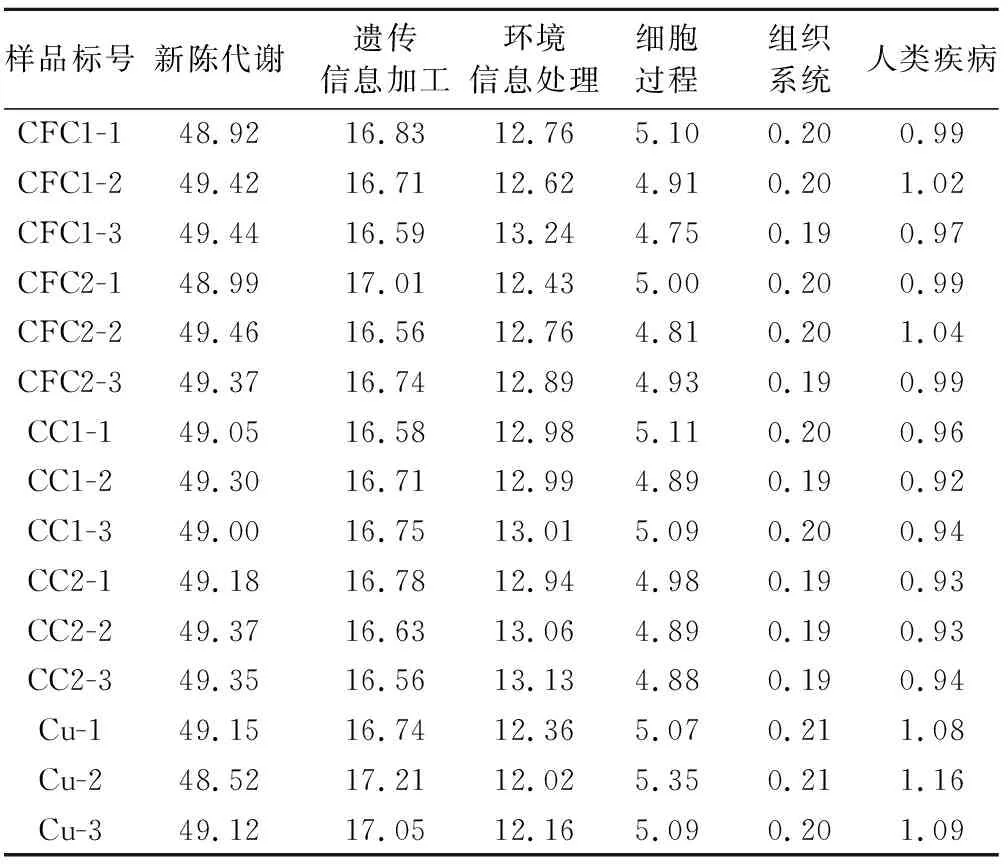
表4 各试验组样品一级功能预测基因的分布 单位:%
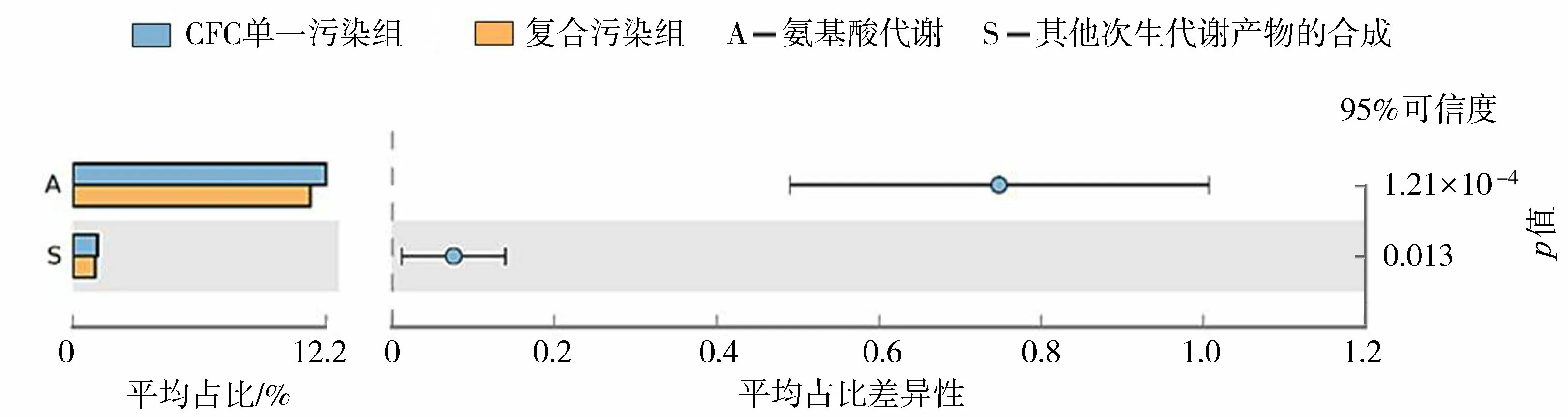
图4 每个样品中二级功能预测基因的显著性差异分析Fig.4 Significance difference analysis of secondary function predicted genes in each sample
2.2 CFC和Cu复合污染下微生物群落与ARGs的交互关系
基于ARGs、MGEs和微生物群落的结构组成,采用网络分析建立了ARGs、MGEs和门水平微生物群落的非随机共发生模式,结果如图5所示,共有23个节点,65条边。整合子intI1是ARGs水平转移的重要遗传元件,网络分析结果显示,intI1是“度”最大的点,与tetA、tetT和sul1 3种ARGs相连,呈现正相关关系,表明intI1在四环素类抗性基因和磺胺类抗性基因的水平转移中可能起到重要作用。该结果与已有研究结果类似:Chen等[33]在对长江口抗生素抗性基因分布特征的研究中发现,intI1与磺胺类抗性基因存在较强的相关关系;王永强[34]研究表明,艾比湖表层水和沉积物中intI1与sul1具有显著的正相关性(p<0.01),说明intI1可能携带sul1基因在环境介质中进行水平迁移,从而增加了环境微生物的耐药性。除此之外,intI1与降氨酸菌门(Aminicenantes)、绿菌门(Chlorobi)、匿杆菌门(Latescibacteria)、芽单胞菌门(Gemmatimonadetes)、硝棘菌门(Nitrospinae)、硝化螺杆菌门(Nitrospirae)、拟杆菌门(Bacteroidetes)和Ignavibacteriae 8个菌群相连,除拟杆菌门和芽单胞菌门外,均呈现正相关关系。同时,intI1和tetT与硝化螺杆菌门、拟杆菌门等均存在正相关性,这可能是intI1和tetT具有相同的宿主菌,intI1携带tetT可能在宿主菌中具有更强的转移性。Lu等[35]同样发现tetX和sul1与短波单胞菌属(Brevundimonas)、苯基杆菌属(Phenylobacterium)和鞘氨醇单胞菌属(Sphingomonas)呈正相关性,表明这些细菌可能是四环素耐药基因和磺胺耐药基因的潜在宿主。同样作为MGEs的插入序列,IS26与QnrA、QnrC、tetA和tetT等多种ARGs相连,没有和微生物相关的节点相连,说明IS26可能更多地在质粒上承担连接ARGs的工作。
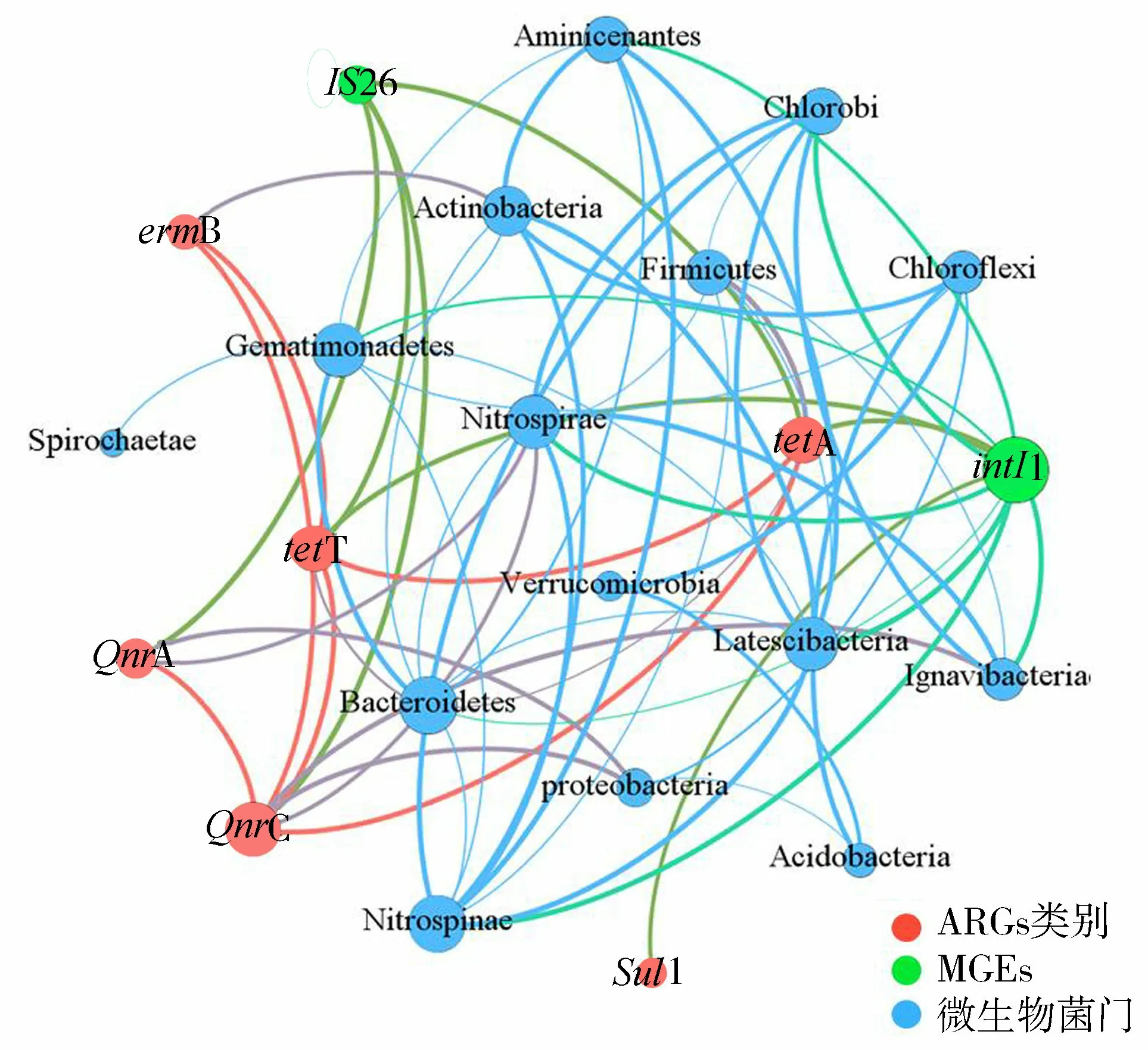
图5 ARGs及其潜在宿主菌分析Fig.5 Analysis of ARGs and their potential host bacteria
从图5还可以看出,氟喹诺酮类抗性基因QnrC与变形菌门、硝化螺杆菌门和Ignavibacteriae相连且均呈正相关关系;四环素类抗性基因tetA与厚壁菌门呈正相关关系,和Ignavibacteriae呈负相关关系;QnrA与变形菌门和硝化螺杆菌门相连,均呈正相关关系;大环内酯类抗性基因ermB与放线菌门存在正相关关系。该结论与Dang等[36]研究结果类似,变形菌门和放线菌门是水环境中门水平的主要ARGs潜在宿主。Tuo等[30]通过网络分析表明,潜在宿主细菌大部分属于放线菌门和厚壁菌门,且intI2在抗性基因传播过程中扮演重要的角色。唐伟欣等[37]对规模化畜禽场粪便中多重耐药菌进行了研究,结果显示,多重耐药菌的菌门集中分布在变形菌门、后壁菌门和放线菌门。Dumas等[38]研究发现,硝化螺旋菌门(Pseudomonas)可能携带多种ARGs,具有多重耐药性,表明这些基因间存在共抗性,或者它们与其潜在宿主之间呈显著正相关关系,从而增强了环境微生物的耐药性。
基于Lefse分析(图6),结果表明在3种污染类型中,CFC单一污染和Cu单一污染试验组在优势菌种上有明显的差异。CFC单一污染试验组的优势菌种多为变形菌门、硝化螺杆菌门和绿菌门的细菌,而Cu单一污染试验组的优势菌种主要为厚壁菌门的细菌。这几个菌门的细菌又恰好是网络分析得出的ARGs潜在宿主,表明各试验组微生物群落中的优势菌种与ARGs有着紧密的联系,潜在宿主的差异可能是造成各试验组中ARGs丰度差异的主要原因。何良英[39]研究发现芽孢杆菌纲(Bacilli)是厚壁菌门下一类重要的细菌菌群,且致病菌Listeriaceae、Streptococcaceae和Staphylococcaceae均属于芽孢杆菌纲。值得注意的是,所属变形菌门和厚壁门的细菌在临床细菌抗性的全球性卫生问题中极其重要,发现了大量抗性基因及其相关MGEs[40]。多种潜在病原微生物通过MGEs进一步传播,将更多的抗性基因带入环境中,从而对生态环境和人类健康带来潜在威胁。
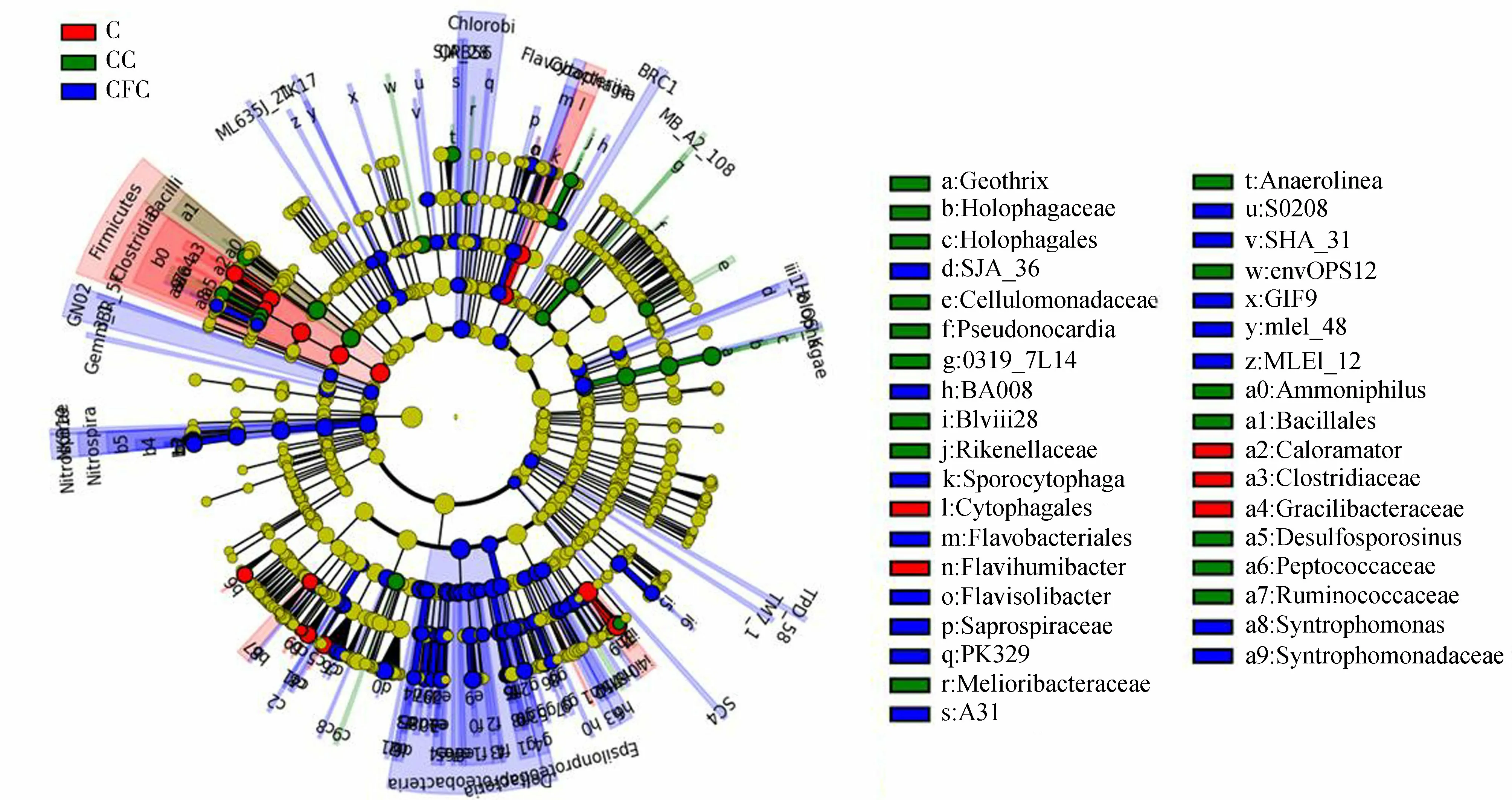
图6 不同污染类型试验组的LEfSe分析Fig.6 LEfSe analysis of experimental groups of different pollution type
2.3 影响ARGs丰度变化的主导因子解析
为了较为全面地分析微生物群落、MGEs以及污染物对ARGs增殖和传播的影响,采用偏冗余性分析对试验结果进行计算(图7)。结果显示94.4%的ARGs丰度变化被所选因素所解释。MGEs对总ARG丰度变化的解释率为32.54%,略高于微生物群落(27.74%),远高于污染物(7.96%)。微生物群落与MGEs和环境因子的联合作用对ARGs丰度变化的总解释率为49.77%,MGEs与其他因子的联合作用对ARGs丰度变化的总解释率为47.65%,本研究室内驯化试验结果与Zhao等[41]的研究结果类似,区别在于本研究微生物群落的总解释率略高于MGEs。造成这一差异的可能原因为:本研究为室内试验,在添加较高浓度污染物的外界压力条件下,微生物群落组成随时间的变化更为显著,进而导致ARGs潜在宿主微生物的丰度显著变化,最终影响ARGs的相对丰度。
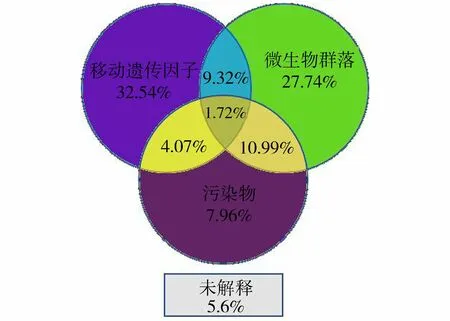
图7 MGEs、微生物群落和污染物对ARGs丰度Fig.7 Interpretation rates for effects of MGEs, bacterial community and pollutants on ARGs abundance
采取路径分析对室内试验涉及的CFC、Cu、微生物群落、MGEs和ARGs按照既定路径进行分析,结果如图8所示。从整体来看,CFC和Cu都通过多个路径影响了ARGs的丰度变化。CFC对微生物群落具有显著的正向影响(p<0.01),对ARGs具有正向影响;而CFC对MGEs具有显著的负面影响(p<0.01);Cu对微生物群落具有负面影响,对MGEs、ARGs具有显著的正向影响(p<0.05);微生物群落对MGEs具有显著的正向影响(p<0.05),对ARGs具有正向影响;MGEs对ARGs具有显著的正向影响(p<0.05)。
路径分析结果表明,受到污染物(CFC和Cu)等环境压力作用的微生物群落影响了MGEs和ARGs,而受到污染物和微生物群落双重影响的MGEs也同样对ARGs的传播有重要的影响。在不同的污染条件下,每一条路径都可能做出相应的响应,如,当CFC成为环境中的主导污染物时,微生物群落可能受到较大的促进作用,一方面可能促进ARGs的垂向扩散过程,另一方面会显著促进MGEs介导的水平转移过程,最终导致ARGs丰度的上升。类似地,Zhou等[42]利用结构方程模型得出水质、微生物群落结构和人类活动的共同作用改变了ARGs分布的结论,其中,河流水体理化参数变化引起微生物群落变化是城市水系ARGs的主要因素。研究结果说明污染物或理化因子更多地通过微生物群落间接地影响ARGs的传播和扩散。
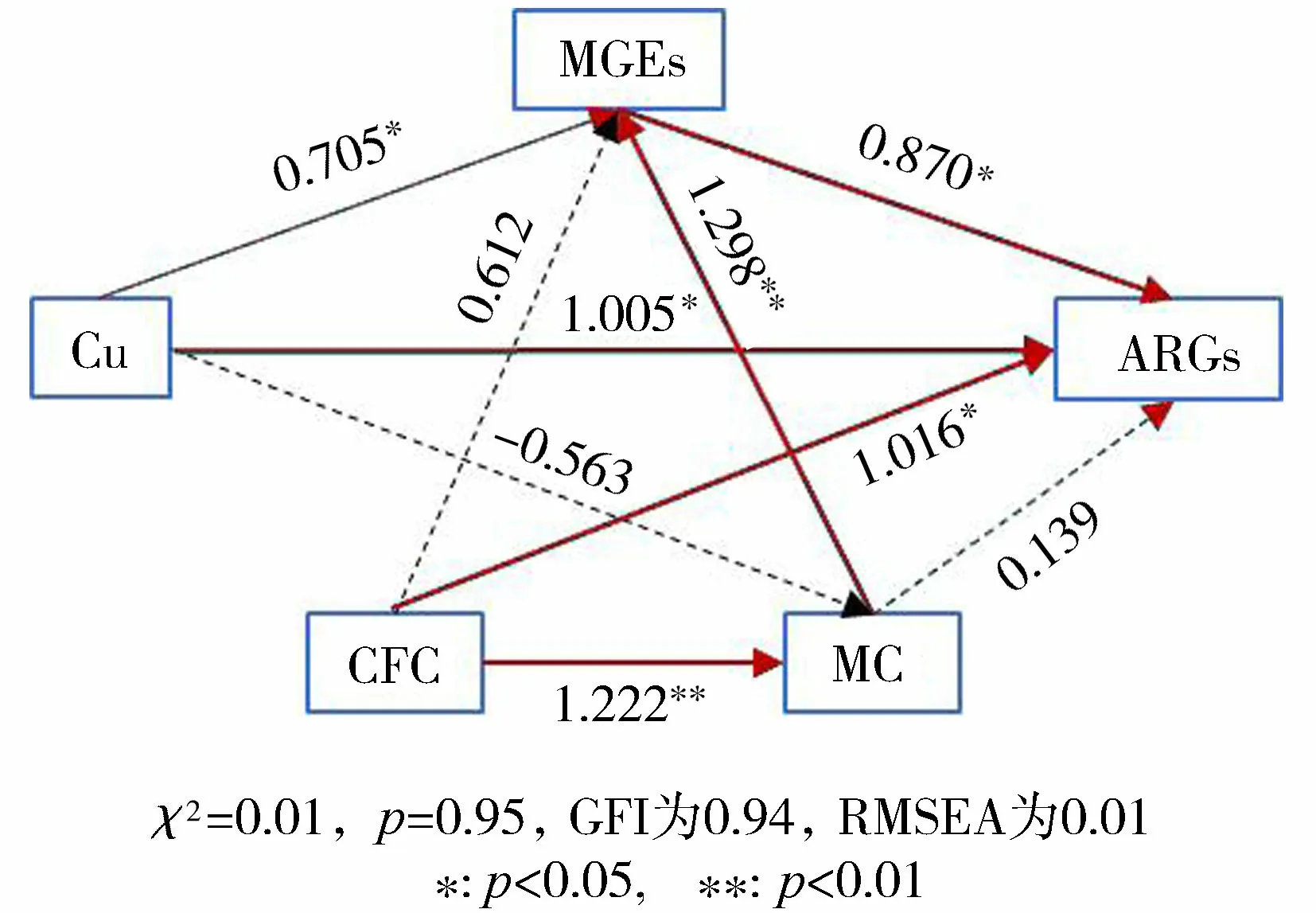
图8 室内试验ARGs与各影响因素的路径分析Fig.8 Path analysis of ARGs and each influencing factor in indoor experiments
3 结 论
a.CFC单一污染、Cu单一污染和复合污染试验组中ARGs丰度、微生物群落组成和部分功能存在显著差异。所有试验组的总ARGs相对丰度呈现出随时间先增后减的变化趋势;变形菌门、酸杆菌门、厚壁菌门、拟杆菌门和绿弯菌门是样品中的优势菌门;与复合污染组相比,氨基酸代谢和其他次生代谢代谢产物的合成使这两个二级功能在CFC单一污染组中有更高的丰度。
b.微生物群落一方面受CFC和Cu等环境因子的影响,另一方面对MGEs和ARGs都具有正向促进作用。共现网络分析表明,intI1与3类ARGs和8个菌群相关,是ARGs和微生物群落之间的桥梁,也是在环境中ARGs传播的重要途径。
c.通过偏冗余分析发现,在MGEs、微生物群落和污染物中,微生物群落是对ARGs丰度变化总解释率最高的因素(49.77%);基于路径分析表明,CFC和Cu等环境因子主要通过微生物群落间接地影响环境中ARGs的增殖与传播。
猜你喜欢
水生生物学报(2022年6期)2022-07-08 09:31:56
海洋通报(2022年2期)2022-06-30 06:06:28
海洋通报(2021年1期)2021-07-23 01:55:24
空间科学学报(2021年1期)2021-05-22 01:36:34
中国比较医学杂志(2020年4期)2020-05-26 05:47:22
水生生物学报(2019年4期)2019-07-20 08:08:10
生物安全学报(2019年3期)2019-02-15 16:54:12
川北医学院学报(2019年6期)2019-02-10 10:48:32
环境保护与循环经济(2017年5期)2018-01-22 02:56:44
海洋渔业(2017年5期)2017-11-07 02:34:58
