
基于QuEChERS-高效液相色谱-串联质谱法检测八角中莽草毒素的研究
2022-11-18 10:07:26钟玉心钱振杰陈彦宏何咏欣杨嘉鑫威龚海锟徐振林
分析测试学报 2022年11期
钟玉心,王 宇,钱振杰,陈彦宏,张 辉,王 伟,何咏欣,杨嘉鑫威,龚海锟,徐振林
(1.华南农业大学 食品学院 广东省食品质量安全重点实验室,广东 广州 510640;2.广州市食品检验所,广东 广州 511405;3.黄埔海关技术中心,广东 广州 510700)
八角(Illicium verum Hook.f),也称八角茴香(Illicium verum),为八角属八角种植物,其种植历史悠久,用途广泛,在亚洲地区常用作烹饪调料[1],因具有镇痛和镇静功能,通常被当作中药用于治疗呕吐、胃痛[2]。同属多种植物如莽草、野八角、日本莽草与八角形态相似,但均含有莽草毒素(Anisatin)(结构式见图1),因此具有不同程度的毒性,误用会引起中毒[3]。莽草毒素属于倍半萜内酯神经毒素,其半数致死量(LD50)为0.76 mg/kg[4]。在欧美地区,因饮用混淆或掺假了日本莽草(Illicium anisatum L.)的八角茴香茶引起的中毒事件时有报道[5]。在中国也发生过误服莽草而导致的中毒事件[6]。由于八角与其伪品莽草等同科植物的外观形态非常相似,仅从完整干果外观难以将其准确区分;对于缺乏形态特征的八角茴香粉,更无法通过外形鉴别真伪,导致八角产品易因原料误掺莽草而引发中毒事件。因此,建立一种准确的八角掺伪鉴别和质量控制方法显得尤为重要。

图1 莽草毒素化学结构式Fig.1 Chemical structure of anisatin
目前,已有研究报道八角茴香及其伪品的鉴别方法,如采用形态学分析方法,通过荧光显微镜、扫描电子显微镜对八角及其伪品进行显微结构鉴别[4,7];采用顶空固相萃取-气相色谱-质谱法比较八角茴香及其伪品中的挥发性风味成分种类与含量的差异[6];基于八角茴香与伪品在毒性成分莽草毒素含量上的差异[8],采用高效液相色谱-串联质谱法(HPLC-MS/MS)对引起中毒的特征成分莽草毒素进行定量分析[4]。其中,HPLC-MS/MS是最常用的八角伪品毒性成分检测方法,可在中毒事故发生后早诊断早干预,降低莽草毒素中毒的死亡率。目前中国药典仅规定了八角中反式茴香脑的含量[9],而对莽草毒素尚未指定标准检测方法。现有的HPLC-MS/MS法主要采用液液萃取、固相萃取的前处理方式,耗时较长。QuEChERS前处理技术具有快速、简单、廉价、高效、安全的优点,已广泛应用于农药残留分析,在生物毒素领域也开始得到应用[10]。本文基于QuEChERS前处理方法,建立了八角中莽草毒素的HPLC-MS/MS检测方法,并用于市售八角茴香中莽草毒素含量的检测。
1 实验部分
1.1 材料与试剂
莽草毒素标准品(CAS:5230-87-5,纯度97.4%)、甲酸(色谱纯)(美国Sigma Aldrich公司);甲醇、乙腈(LC/MS级,美国Thermo公司);乙酸(色谱纯,天津科密欧化学试剂公司);QuEChERS盐包(4.0 g无水硫酸镁,1.0 g氯化钠)、乙酸缓冲盐萃取盐包(6.0 g无水硫酸镁,1.5 g乙酸钠)、柠檬酸缓冲盐萃取盐包(4.0 g无水硫酸镁,1.0 g氯化钠,1.0 g二水合柠檬酸钠,0.5 g柠檬酸氢二钠)均购于美国Agilent公司;N-丙基乙二胺(PSA)、C18和石墨化碳黑(GCB)均购于上海安谱实验科技股份有限公司。
实验所用八角样品均为市售。
1.2 仪器与设备
高效液相色谱-串联质谱仪(AB Sciex 5500 QTRAP,美国AB SCIEX公司);电子天平(CP225D,德国Sartorius公司);Milli-Q Academic超纯水系统(德国Millipore公司);高速冷冻离心机(Sorvall ST 16R,美国Thermo公司);多位点涡旋振荡器(Multi Reax,德国Heidolph公司);圆周振荡器(MA 3 basic,德国IKA公司);Accucore aQ C18色谱柱(2.1 mm×150 mm,2.6 μm,美国Thermo公司)。
1.3 标准溶液的配制
莽草毒素标准储备液(1.0 mg/mL):准确称取莽草毒素标准品10 mg(精确至0.01 mg),以甲醇溶解后,转移至10 mL容量瓶中,用甲醇定容至刻度,于-20℃保存。
莽草毒素标准中间液(1.0 μg/mL):准确吸取100 μL莽草毒素标准储备液至100 mL容量瓶中,用乙腈定容至刻度,于-20℃保存。
莽草毒素标准工作液:分别移取适量莽草毒素标准中间液,用乙腈稀释定容,配制成1.0、5.0、10.0、20.0、50.0、100.0 ng/mL的标准工作液,现用现配。
1.4 样品前处理
称取0.5 g样品(精确至0.01 g)至50 mL具塞离心管中,加入1颗陶瓷均质子、5 mL超纯水、10 mL乙腈,涡旋提取10 min,加入乙酸缓冲盐萃取盐包,盖上离心管盖,剧烈振荡1 min,以8 000 r/min离心3 min。吸取2 mL上清液至QuEChERS样品净化管中(内含200 mg PSA、50 mg GCB),涡旋1 min,以8 000 r/min离心3 min,过0.22 μm有机微孔滤膜,供HPLC-MS/MS检测。
1.5 分析条件
1.5.1 色谱条件色谱柱:Accucore aQ C18柱(2.1 mm×150 mm,2.6 μm);流动相:A为甲醇,B为含0.1%甲酸的水溶液;流速:0.3 mL/min;进样量:2 μL;柱温:40℃;梯度洗脱程序:0~4.0 min,95% B;4.0~7.0 min,95%~20% B;7.0~9.0 min,20% B;9.0~9.1 min,20%~95% B;9.1~12.0 min,95%B。
1.5.2 质谱条件离子源:电喷雾离子源;离子扫描模式:负离子模式(ESI-);扫描方式:多反应监测(MRM)模式;离子源温度:500℃;气帘气:206.8 kPa(30 psi);离子化电压:4 500 V;碰撞气:55.2 kPa(8 psi);辅助加热器1(GS1):344.7 kPa(50 psi);辅助加热器2(GS2):344.7 kPa(50 psi)。
1.6 数据处理
采用AB Sciex Analyst Software软件进行数据采集,Multi Quant(3.0.2)软件处理数据,Origin 2019b软件作图。
2 结果与讨论
2.1 质谱条件优化
以甲醇-水(1∶1)为溶剂,配制100 ng/mL的莽草毒素标准溶液,注入质谱系统,进行正负离子扫描,筛选母离子。结果表明:负离子模式下分子离子[M-H]-(m/z327.1)的响应值最高。在-5~-50 eV范围内逐步改变碰撞能量,直至母离子强度为基峰强度的1/3,筛选出m/z297.1、126.9的子离子,组建MRM离子对,并优化去簇电压及碰撞能量。质谱参数见表1。

表1 莽草毒素质谱参数Table 1 Mass spectrometric parameters for anisatin
2.2 流动相优化
考察了普通C18色谱柱和aQ柱对莽草毒素的分离效果,结果表明,两者均可对莽草毒素进行测定,但不同色谱柱上的峰形差别较大。莽草毒素在aQ色谱柱上的峰形尖锐,分离效果好,故选择aQ柱作为分析柱。考察了乙腈-水、甲醇-水、甲醇-0.1%甲酸水溶液作为流动相的效果。结果表明,以乙腈-水为流动相的分离效果较差,存在峰重叠现象,调节梯度洗脱程序也无法得到满意的分离效果;而甲醇极性弱于乙腈,能实现目标化合物和杂质的分离;加入0.1%甲酸虽抑制了莽草毒素电离导致响应有所降低,但可减少基质效应的影响。故选择甲醇-0.1%甲酸水溶液为流动相。在优化洗脱程序下,莽草毒素标准溶液与加标样品的色谱图见图2。

图2 莽草毒素标准溶液(A)与加标样品(B)的色谱图Fig.2 Chromatograms of anisatin standard solution(A)and a spiked sample(B)
2.3 样品前处理方法优化
2.3.1 提取溶剂的选择QuEChERS常用的提取溶剂为甲醇、乙腈、丙酮、乙酸乙酯等。相较于其它提取溶剂,乙腈具有很好的破壁作用及无机盐盐析作用,加入无机盐可使乙腈与水相分层,提高提取效率[11-12]。本实验首先考察了乙腈为提取溶剂时的提取效率;同时考察了加入1%乙酸对提取效果的影响。结果显示,乙腈的提取率达97%,而1%乙酸的加入对提取率的影响不明显,最终选择乙腈作为提取溶剂。
2.3.2 缓冲盐体系的选择本实验比较了普通QuEChERS盐包(1.0 g氯化钠,4.0 g无水硫酸镁)、乙酸缓冲盐萃取盐包(6.0 g无水硫酸镁,1.5 g乙酸钠)、柠檬酸缓冲盐萃取盐包(4.0 g无水硫酸镁,1.0 g氯化钠,1.0 g二水合柠檬酸钠,0.5 g柠檬酸氢二钠)、氯化钠对提取效果的影响。结果表明,使用普通QuEChERS盐包及氯化钠作为盐析剂时,回收率分别为92.9%及94.4%;使用柠檬酸缓冲盐萃取盐包的回收率最低,仅为77.7%;而使用乙酸缓冲盐萃取盐包的回收率可达98.0%。因此本实验采用乙酸缓冲盐萃取体系。
2.3.3 净化剂及其用量的选择八角中含有大量的反式茴香醚及莽草酸[13],必须进行净化,避免杂质干扰检测、污染质谱系统。QuEChERS方法常用的净化剂有PSA、C18、GCB[12]。PSA可以消除样品中的有机酸、色素、糖类、脂肪酸等杂质[14],C18可去除油脂和弱极性杂质[15-16],GCB可去除色素及平面结构物质[17-18]。因此,考察了上述3种净化剂对莽草毒素回收率的影响(图3)。结果显示,C18对莽草毒素的吸附作用最大,其平均回收率仅为64%;而随着PSA用量的增加,莽草毒素的回收率不断提高,PSA用量为200 mg时与500 mg的回收率相近,因此选择200 mg PSA作为净化剂;GCB虽对莽草毒素有一定的吸附,但其对色素的净化效果较佳,经GCB净化后的提取液最澄清。因此考察了PSA与GCB复合净化的回收率。在综合考虑回收率、色素净化效果与成本的基础上,200 mg PSA+50 mg GCB可取得最佳的净化效果。

图3 不同净化剂对莽草毒素回收率的影响(n=6)Fig.3 Effect of different purificants on anisatin recoveries(n=6)
2.4 方法学评价
2.4.1 基质效应的评价八角中反式茴香醚、有机酸、色素等杂质的存在对莽草毒素的检测存在干扰。本实验将莽草毒素配制在乙腈中得到样品A;对八角样品进行前处理得到样品B;对八角样品进行前处理后加入标准溶液得到样品C,对上述3个样品进行同时检测,并分别以A、B、C作为其回收率值考察基质效应(ME)。
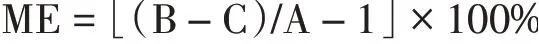
当ME为正值,表示存在基质增强效应;当ME为负值,表示存在基质抑制效应。ME的绝对值大于50%为强基质效应,20%~50%为中等基质效应,而小于20%则为弱基质效应[19-21]。结果表明,莽草毒素经本方法处理后存在弱的基质抑制效应(-8.6%),对结果影响较小。
2.4.2线性范围与检出限以乙腈为溶剂,配制1.0、5.0、10.0、20.0、50.0、100.0 ng/mL质量浓度的标准溶液,在优化条件下进行测定,以质量浓度为横坐标(x),定量离子对的峰面积为纵坐标(y)绘制标准曲线,得到线性方程为y=2 029.358 55x+133.657 11。结果显示,莽草毒素在1.0~100.0 ng/mL质量浓度范围内呈现良好的线性关系,相关系数(r2)达到0.999 99。分别以3倍和10倍信噪比(S/N)确定方法的检出限(LOD)和定量下限(LOQ)[22],莽草毒素的LOD和LOQ分别为0.006 mg/kg和0.02 mg/kg。与现行方法相比,本方法具有操作简单、快速和定量准确的优点[1,4]。
2.4.3 回收率与相对标准偏差以八角粉为基质,添加0.02、0.20、0.40、1.00 mg/kg 4个水平的莽草毒素标准品,每个水平进行6次平行实验,考察回收率与相对标准偏差(RSD),结果见表2。由表2可知,莽草毒素的平均回收率为92.5%~112%,RSD为1.1%~6.0%,方法准确度和精密度满足GB/T 27404-2008[23]的要求,可应用于八角中莽草毒素的分析检测。

表2 莽草毒素的回收率及相对标准偏差(n=6)Table 2 Recoveries and relative standard deviations of anisatin(n=6)
2.5 实际样品检测
采用本方法对市售的10份八角样品进行测定,其中2份样品莽草毒素的含量小于0.02 mg/kg,7份含量为0.02~0.30 mg/kg,1份含量为2.96 mg/kg。由于莽草毒素含量大于1.00 mg/kg应考虑存在掺假可能[8],因此市售八角存在潜在的掺假问题,有一定的安全风险。
3 结论
本研究建立了一种八角中莽草毒素的QuEChERS-高效液相色谱-串联质谱检测方法。该方法前处理简单快捷,方法学指标满足检测要求,适用于八角中莽草毒素的含量检测及八角的真伪鉴别,在八角的食品安全监管和风险监控方面具有一定的应用价值。
猜你喜欢
中国乡村医药(2023年22期)2023-11-30 06:35:26
煤化工(2022年3期)2022-07-08 07:24:42
考试与评价·高二版(2021年3期)2021-09-10 07:22:44
数学物理学报(2020年5期)2020-11-26 06:06:28
养生保健指南(2019年10期)2019-12-16 01:46:38
天然产物研究与开发(2018年8期)2018-09-10 05:48:38
恋爱婚姻家庭·养生版(2018年12期)2018-01-15 02:25:26
公民与法治(2016年14期)2016-05-17 04:15:03
中国资源综合利用(2016年10期)2016-01-22 08:36:09
家庭医药·快乐养生(2015年7期)2015-09-10 07:22:44
