
重铬酸钾和高锰酸钾电位落差法测定砂岩型铀矿氧化还原电位的探讨
2022-11-11 03:32王娜王家松曾江萍李强吴磊陈枫
岩矿测试 2022年5期
王娜, 王家松, 曾江萍, 李强, 吴磊, 陈枫
(1.中国地质调查局天津地质调查中心, 天津 300170;2.中国地质调查局泥质海岸带地质环境重点实验室, 天津 300170;3.核工业二〇三研究所, 陕西 咸阳 710086)
铀矿作为国家战略资源和能源矿产,在国防建设和经济发展中发挥着重要作用,而砂岩型铀矿在铀矿中的占比最大。中国砂岩型铀矿主要分布在北方大型沉积盆地,如新疆伊利盆地、准格尔盆地、塔里木盆地、内蒙古鄂尔多斯北部、二连盆地、东北松辽盆地等[1-7],随着地浸技术的应用,砂岩型铀矿成为铀矿勘查的主攻方向[8-11]。铀是氧化还原敏感元素,在氧化环境中主要以六价态的碳酸铀酰络合物[UO2(CO3)3]4-和[UO2(CO3)3]6-存在;在氧化还原过渡带,两种碳酸盐铀酰络合物[UO2(CO3)3]4-和[UO2(CO3)3]6-降低分解,[UO2]2+处于饱和状态;在还原环境中以四价态的沥青铀矿(UO2)形式存在[12-15]。四价铀较稳定,易富集成矿;六价铀溶解度较高,易于迁移。当岩石的氧化还原电位较高时,流体从岩石中浸取铀;当氧化还原电位较低时,液体中的铀可能被还原沉淀从而形成铀矿。因此,氧化还原电位对铀矿矿体的圈定具有指示意义[16-20]。
氧化还原电位是由氧化还原电对和标准氢电极组成原电池,在电极反应达到平衡时的电动势(Eh)[21],反映了整个体系氧化性或还原性强弱的相对程度[22-24]。氧化还原电位的测定分为直接测定法和电位落差法(差减电位法)[12,25]。直接测定法是采用电位仪或者去极化仪等仪器测定样品的电动势。电位落差法是测定加入含有还原组分的样品后该氧化剂溶液的氧化还原电位变化值(ΔEh)。张卫民等[14]采用直接测定法测定新疆伊犁盆地512铀矿床样品,利用测量的大量数据划分和确定了砂岩型铀矿层间氧化带中的氧化带、氧化还原过渡带和还原带的大体位置。但直接测定法操作繁琐,干扰因素多,易受空气中氧气的影响,所测电位值取决于样品中水溶性物质的多少,不能真实地反映岩石的氧化还原电位值[24-25];电位落差法可兼顾水溶性和不溶于水的还原组分,能够客观地反映样品的还原能力,同时氧化剂的存在可忽略氧气的影响[12,25]。王剑锋[26]利用高锰酸钾落差法测定的氧化还原电位进行地球化学分带,以此研究了铀矿成矿作用机理和矿化富集规律,但是并未将高锰酸钾落差法实验条件加以优化细化。
重铬酸钾和高锰酸钾作为两种电位值稳定的强氧化剂,本文采用重铬酸钾和高锰酸钾电位落差法测定砂岩型铀矿的氧化还原电位,探讨了溶液介质和氧化剂浓度、平衡电位和浸泡时间、样品与氧化剂溶液的固液比对ΔEh测定的影响,确定了两种方法最佳测量条件,通过测定8个砂岩型铀矿样品,建立了砂岩型铀矿氧化还原电位测定的分析方法,对砂岩型铀矿氧化还原分带的划分、成矿机理和矿产分布规律的研究提供了支撑。
1 实验部分
1.1 仪器与主要试剂
雷磁pHS-25型氧化还原电位测定仪(上海仪
电科学仪器有限公司):工作电极(213-01铂电极);参比电极(232甘汞电极)。
重铬酸钾为基准试剂(天津科瑞恩化工销售有限公司),高锰酸钾(天津渤化化学试剂有限公司)。其余试剂均为分析纯(天津市致远化学试剂有限公司)。实验用水为二次去离子水。
1.2 试剂配制
硫酸亚铁铵-硫酸铁铵标准溶液:称取39.21g硫酸亚铁铵和48.22g硫酸铁铵于烧杯中,加入56mL浓硫酸,搅拌后,将悬浊液倒入含600mL蒸馏水的1000mL烧杯中,放置两天,待澄清后转移至1000mL容量瓶中并定容至刻度。
0.10mol/L的1/6K2Cr2O7溶液:称取24.51g重铬酸钾于烧杯中溶解,转移至5000mL容量瓶中,加入1000mL 50%硫酸,定容至刻度并摇匀。
0.10mol/L的1/3KMnO4溶液:称取5.27g高锰酸钾溶于1000mL水中,加热煮沸,放置过夜,用砂漏斗或者脱脂棉过滤至1000mL棕色容量瓶中,加入20mL 10%氢氧化钾,用煮沸放至常温的去离子水定容至刻度并摇匀。
1.3 实验方法
(1)样品处理:实验所测试的样品(样品编号为样品1~3、Q001和L002~L009)均为采自鄂尔多斯盆地的灰绿色砂岩岩心样品,经过粗碎、中碎、细碎三个阶段,每个阶段包括破碎、过筛、混匀和缩分四道工序,粉碎至150~200目。样品缩分公式、破碎粒径、质量检查等具体要求参照《地质矿产实验室测试质量管理规范 第2部分:岩石矿物分析样品制备》(DZ/T 0130.2)。
(2)电极预处理:铂电极依次在纯水、无水乙醇、0.1mol/L盐酸中超声处理3min后,再浸泡于0.1mol/L盐酸中60min,用去离子水洗净;参比电极在使用前浸泡于0.10mol/L盐酸中60min,用去离子水洗净。
(3)样品测定:将经预处理的铂电极和参比电极插入重铬酸钾溶液或高锰酸钾溶液中,金属表面便会产生电子转移,电极与溶液产生电位差,电极反应达到平衡时,记录相对于参比电极的电位值Eh1。称取粉碎至150~200目的样品2.50g于150mL瓶中,加入50mL重铬酸钾溶液或者高锰酸钾溶液,摇匀后盖紧,每30min摇匀一次,1h后测定高锰酸钾溶液上清液电位值Eh2,2h后测定重铬酸钾上清液电位值Eh2。氧化还原电位值ΔEh=Eh1-Eh2。
2 结果与讨论
2.1 重铬酸钾法溶液介质和氧化剂浓度的选择

氧化剂浓度过低时,外界因素如光线、灰尘及空气中的还原颗粒对其稳定性影响较大[29];浓度过高时,仪器灵敏度和测量稳定性降低。合理的氧化剂浓度既可以降低分析误差,又能保证较大的落差电位。按照1.3节所示的实验方法测量不同浓度重铬酸钾的Eh及砂岩型铀矿-重铬酸钾浸泡液的ΔEh,测量结果如表1所示,1/6K2Cr2O7溶液浓度在0.05~0.15mol/L内,Eh值随着浓度增加而升高,在0.20mol/L时Eh有所下降。当1/6K2Cr2O7溶液浓度为0.01mol/L时,氧化能力弱于2.5g砂岩型铀矿样品的还原能力,氧化剂不足而被完全还原;浓度为0.10mol/L和0.15mol/L时,氧化能力强于样品的还原能力,砂岩型铀矿样品被完全氧化,ΔEh值高于浓度为0.05mol/L和0.20mol/L时的ΔEh值。根据表1的数据,选取氧化剂1/6K2Cr2O7溶液浓度0.10mol/L。

表1 不同浓度重铬酸钾条件下重铬酸钾及样品-重铬酸钾浸泡液的电位值
2.2 高锰酸钾法溶液介质和氧化剂浓度的选择
高锰酸钾法测定氧化还原电位是在pH=11~13的碱性介质中进行[26],当砂岩型铀矿样品中的还原组分被氧化,Mn7+被还原为MnO2时,溶液的电位值降低。两种电位落差法测定氧化还原电位时,1/3mol KMnO4与1/6mol K2Cr2O7转移电子数相同,为了直观地比较两种方法测量砂岩型铀矿的氧化还原电位值,选取1/3KMnO4溶液的浓度与1/6K2Cr2O7相同,均为0.10mol/L。
按照1.3节的实验方法考察了氢氧化钾不同浓度下高锰酸钾溶液及砂岩型铀矿高锰酸钾浸泡液的Eh值,结果如表2所示,氢氧化钾浓度在0.0~1.5×10-3时,高锰酸钾溶液Eh逐渐降低,但在氢氧化钾浓度为1.5×10-3~10.0×10-3时趋于稳定。加入砂岩型铀矿后,氢氧化钾浓度在0.0~1.0×10-3时ΔEh随氢氧化钾浓度增加而增大,并在氢氧化钾浓度为1.0×10-3~10.0×10-3时趋于稳定。为保证溶液的pH=11~13及ΔEh的稳定,选取高锰酸钾法中的氢氧化钾介质浓度为2.0×10-3。
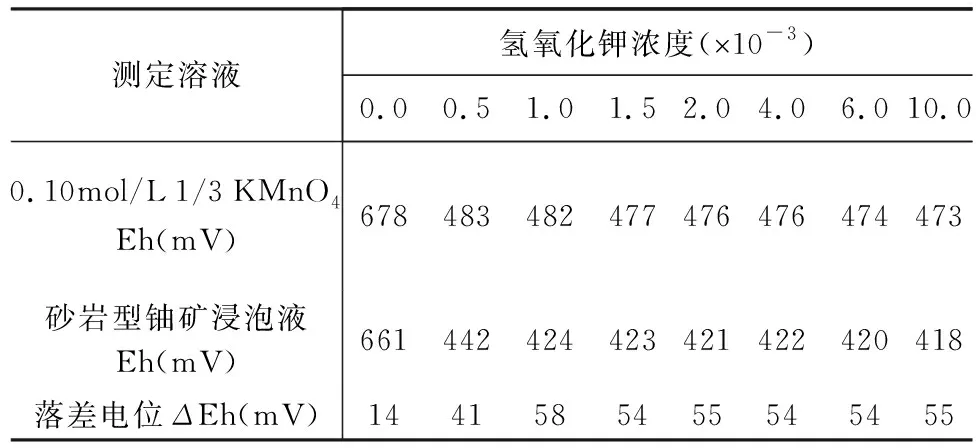
表2 不同浓度氢氧化钾条件下高锰酸钾及样品-高锰酸钾浸泡液的电位值
2.3 两种方法平衡电位时间、样品浸泡时间及固液比的选择
平衡电位时间是影响电位值测量准确度的重要因素[29]。一般是将某一时间点的稳定电位称为相对平衡电位[30]。实验考察了硫酸亚铁铵-硫酸铁铵标准溶液、重铬酸钾溶液(10%硫酸介质的0.10mol/L 1/6K2Cr2O7溶液)、砂岩型铀矿重铬酸钾浸泡液(10%硫酸介质的2.5g样品-50mL0.10mol/L1/6K2Cr2O7溶液)、高锰酸钾溶液(2.0×10-3氢氧化钾介质的0.10mol/L1/3KMnO4溶液)和砂岩型铀矿高锰酸钾浸泡液(2.0×10-3氢氧化钾介质的2.5g样品-50mL 0.10mol/L1/3KMnO4溶液)不同时间下电位值。
测定结果如表3所示,硫酸亚铁铵-硫酸铁铵标准溶液、高锰酸钾溶液和砂岩型铀矿高锰酸钾浸泡液在5min时可达到相对平衡电位,而重铬酸钾溶液和砂岩型铀矿重铬酸钾浸泡液在15min时达到相对平衡电位。这是由于硫酸亚铁铵-硫酸铁铵标准溶液和高锰酸钾溶液属于强平衡体系,短时间内可达到稳定电位值;而重铬酸钾溶液属于弱平衡体系,需较长时间达到平衡电位。因此,测量电位前使用硫酸亚铁铵-硫酸铁铵标准溶液检测氧化还原电位仪的状态;高锰酸钾落差法平衡电位时间选择5min,重铬酸钾落差法选择15min。

表3 不同平衡电位时间下溶液的电位值
浸泡时间直接影响砂岩型铀矿与氧化剂的反应效果,从而影响电位值及电位差。按照1.3节实验方法测量不同浸泡时间下三个样品两种方法的ΔEh值,结果见表4,重铬酸钾法三个样品的ΔEh值在60min后达到最大,高锰酸钾法的ΔEh值在30min即可达到平衡,为了使样品被充分氧化,选取重铬酸钾法样品浸泡时间120min,高锰酸钾法为60min。
砂岩型铀矿样品质量与氧化剂溶液体积的固液比(g∶mL)是影响氧化还原效果的又一重要因素[29-31]。实验考察了固液比分别为0.5∶50、1.0∶50、1.5∶50、2.0∶50、2.5∶50、3.0∶50、5.0∶50时三个砂岩型铀矿样品两种方法的ΔEh值。从表4可以看出,随着固液比的增加,还原性物质增多,两种方法的ΔEh值逐渐升高;但是当固液比大于2.5∶50时,重铬酸钾法样品1和样品3的ΔEh值增幅渐缓,而高锰酸钾法中固液比为1.0∶50时三个样品ΔEh值增幅放缓。为了使样品的还原物质能被充分氧化且通过ΔEh值更好地反映样品还原能力,选择重铬酸钾法固液比为2.5∶50,高锰酸钾法固液比为1.0∶50。
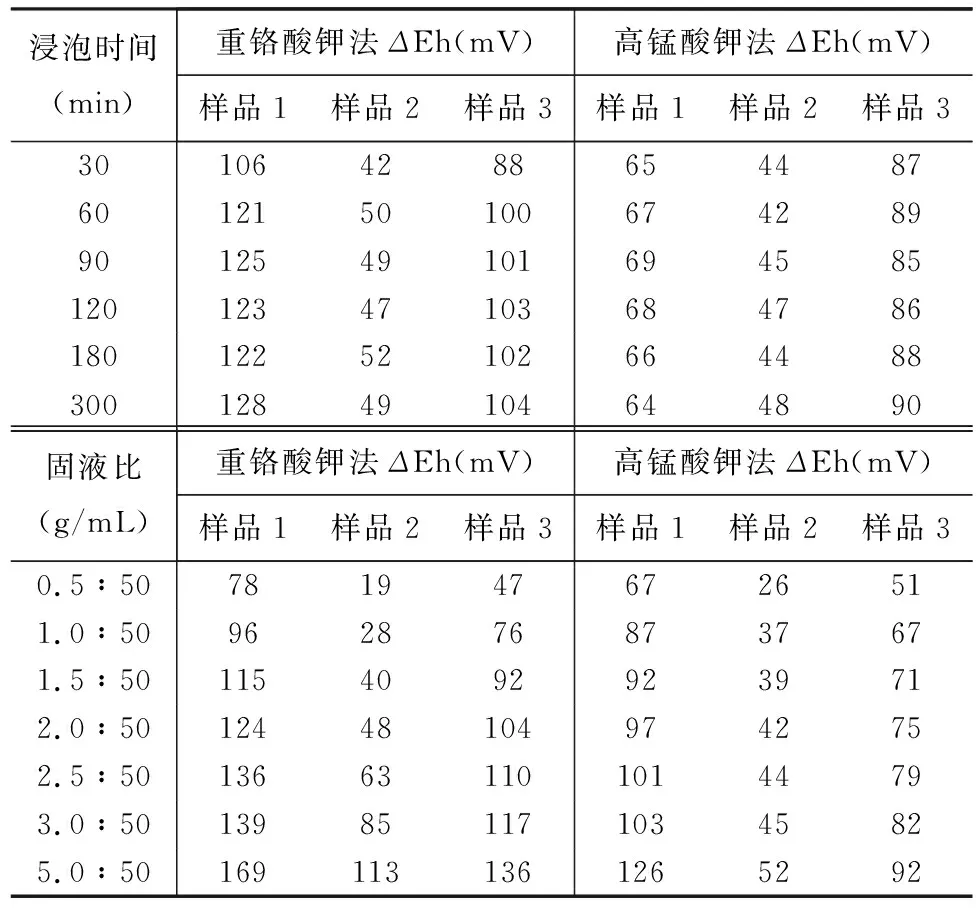
表4 不同浸泡时间和固液比下两种方法的ΔEh值
2.4 两种方法的测试结果
按照优化后的重铬酸钾电位落差法(2.5g砂岩型铀矿样品于10%硫酸介质的50mL 0.10mol/L氧化剂1/6K2Cr2O7溶液中浸泡120min,平衡电位时间15min)和高锰酸钾电位落差法(1.0g砂岩型铀矿样品于2.0×10-3氢氧化钾介质的50mL 0.10mol/L氧化剂1/6KMnO4溶液中浸泡60min,平衡电位时间5min)测定ΔEh值,具体分析结果见表5。8个样品重铬酸钾法的ΔEh值在15~118mV之间,测量10次的RSD为2.50%~7.44%;高锰酸钾法的ΔEh值在45~89mV之间,测量10次的RSD为0.89%~1.42%。从分析数据来看,8个样品重铬酸钾法的ΔEh值更分散、跨度更大,更能直观地看出不同样品还原能力的差别,但ΔEh稳定性要低于高锰酸钾法。虽然两种方法的ΔEh在数值上并不相等,但二者表现出较强的线性相关性,高锰酸钾法ΔEh值和重铬酸钾法ΔEh值的线性方程为y=0.3882x+40.07,相关系数为0.9882,说明两种方法在测量8个砂岩型铀矿样品ΔEh的相对水平方面具有一致性。

表5 两种方法测量砂岩型铀矿样品的ΔEh值
2.5 砂岩型铀矿氧化还原环境和氧化还原分带的判断
矿物氧化系数法可以判断矿物的氧化还原环境,对揭示矿床形成机理及普查找矿具有重要意义。通常用矿物中Fe2O3/FeO质量的百分比或者分子量百分比或分子式中不同价态的Fe3+/Fe2+比值来表示[32-33]。若Fe3+/Fe2+≫1,说明矿样中以Fe3+为主,是氧化环境;若Fe3+/Fe2+>1,矿样中Fe3+略大于Fe2+,是弱氧化环境;若Fe3+/Fe2+<1,矿样中Fe2+稍大于Fe3+,是弱还原环境;若Fe3+/Fe2+≪1,说明矿物中以Fe2+为主,是还原环境。
根据ΔEh值也可对矿物进行氧化还原分带,判断岩性的氧化还原容量[25-26]。按照文献报道,ΔEh<25mV时为氧化岩石带;ΔEh在25~45mV时为氧化还原过渡带;ΔEh在45~65mV时为还原岩石带;ΔEh>65mV时为强还原带。
采用重铬酸钾滴定法[34]及熔片法[35-36]分别测定样品Q001、L003、L006和L008中Fe2+和Fe3+的含量,计算Fe3+/Fe2+比值,判断矿物氧化还原环境,同样按照高锰酸钾法测定上述矿样的ΔEh并判断氧化还原分带[26]。结果如表6所示,两种方法判断的氧化还原环境与氧化还原分带基本一致,Q001和L008分别为氧化岩石带和强还原带,氧化系数法判断为氧化环境和还原环境;L003和L006分别为氧化还原过渡带和还原岩石带,对应弱氧化环境和弱还原环境。通过2.4节分析得出,以重铬酸钾和高锰酸钾作为氧化剂的两种电位落差法在测量砂岩型铀样品ΔEh 的相对水平方面一致,重铬酸钾法测定的ΔEh数值更分散、跨度更大,说明该法测定的ΔEh值也可应用于砂岩型铀矿的氧化还原分带。
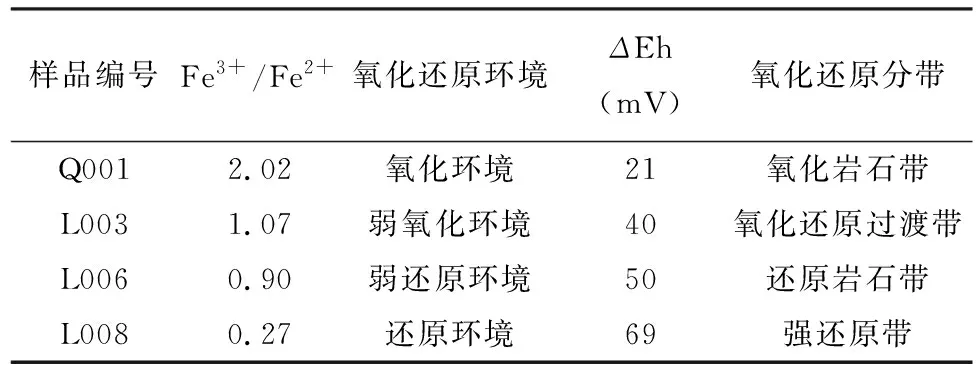
表6 氧化还原环境和高锰酸钾电位落差法氧化还原分带的判断
3 结论
采用重铬酸钾法和高锰酸钾法两种电位落差法测定砂岩型铀矿的氧化还原电位,系统研究了影响砂岩型铀矿ΔEh测量的因素,包括介质浓度、氧化剂浓度、平衡电位时间、样品浸泡时间及固液比的选取,利用优化后的两种方法测量8个砂岩型铀矿样品的ΔEh值,二者在测量氧化还原电位的相对水平方面具有一致性。采用高锰酸钾法测量的ΔEh值判断砂岩型铀矿氧化还原分带,与氧化系数法判断的矿物氧化还原环境吻合,两种电位落差法测量的ΔEh值可用于砂岩型铀矿氧化还原分带的划分。
两种落差电位法能准确快速地测量砂岩型铀矿的ΔEh值,有效地解决了直接电位法操作繁琐、影响因素多和测量误差大等问题,为其他岩石、土壤、矿物氧化还原电位的测量提供了借鉴,但重铬酸钾电位落差法不适用于测量碳酸盐类矿物的氧化还原电位。另外应用于实际样品测量时,氧化还原电位还需与地质构造、岩性岩相、水文地质和古地理研究等密切结合才能获得可靠成果。
猜你喜欢
天津科技(2022年6期)2022-06-21
四川冶金(2021年6期)2021-02-15
宜春学院学报(2020年9期)2020-12-03
实用临床护理学杂志(电子版)(2020年4期)2020-05-21
中学化学(2019年4期)2019-08-06
中学化学(2019年4期)2019-08-06
浙江农业学报(2017年1期)2017-05-17
林业与生态(2016年2期)2016-02-27
中国资源综合利用(2016年7期)2016-02-03
中国卫生标准管理(2015年6期)2016-01-14
