
核糖体泛素化在心血管疾病中的研究进展
2022-11-10 03:51:28李玉洁张瑜吴诚洁陆琳王燕丽李杨欣
心血管病学进展 2022年10期
李玉洁 张瑜 吴诚洁 陆琳 王燕丽 李杨欣
(苏州大学医学院心血管病研究所 苏州大学附属第一医院心脏大血管外科,江苏 苏州 215123)
目前,心血管疾病(cardiovascular disease,CVD)的死亡人数约占全球总死亡人数的1/3,难以预防且难以治愈,因此,CVD正成为威胁全球人民健康的第一大杀手[1]。CVD是发生于心脏和循环系统的疾病,其发病机制复杂。已有报道称,不同类型的细胞、慢性炎症反应、多种细胞因子和黏附分子都是CVD发病的潜在原因[2]。近期研究表明,核糖体泛素化也参与了CVD的发生发展。现就核糖体泛素化在CVD中的研究进展做一综述,旨在为CVD的治疗提供新思路。
1 核糖体
细胞是生命最小的代谢功能单位,能感知外界信号并作出反应,这一过程受到蛋白质合成的严格控制[3]。核糖体,也称核蛋白体,是细胞中负责蛋白质合成的细胞器,由核糖体RNA(ribosomal RNA,rRNA)和核糖体蛋白(ribosomal proteins,RPs)构成,几乎存在于所有细胞内的细胞器,即使是体积最小的支原体细胞内也含有上百个核糖体[4]。半个世纪以前,人们通过对哺乳动物细胞中的RNA进行放射性脉冲标记,发现真核生物核仁中的核糖体由一个巨大前体rRNA组成,即45S rRNA前体,之后被加工成5.8S、18S和28S rRNA[5]。其后,对脉冲标记的RNA进行蔗糖梯度离心,发现了各种rRNA中间体的前核糖体颗粒(称为43S、66S和90S)[5]。值得注意的是,66S和90S前核糖体颗粒比成熟亚基具有更高的蛋白质/RNA比,据此推测核糖体可能包含其他蛋白质[5]。
多年的研究[6]证明,真核生物核糖体由4种rRNA和80种RPs组成。在从核仁到细胞质的运输过程中,这些前核糖体与它们的大部分非核糖体因子分离,然后成为成熟的40S和60S亚基,用于蛋白质翻译[7]。核糖体60S大亚基,包含5S rRNA、5.8S rRNA、28S rRNA和47种RPs,而40S小亚基包含18S rRNA和33种RPs[6]。组成大、小亚基的RPs分别称之为大亚基核糖体蛋白(ribosomal protein large subunit,RPL)和小亚基核糖体蛋白(ribosomal protein small subunit,RPS)[8]。RPs除了参与蛋白质的合成之外,还能调控细胞生长、增殖、分化、凋亡和DNA修复,被称为核糖体外功能[9](表1)。另外,RPs的功能障碍与CVD的发生发展密切相关[9-10]。

表1 部分RPs的核糖体外功能
2 泛素化
泛素化是指泛素蛋白(ubiquitin,Ub)以共价键附着在底物蛋白上调节蛋白质的降解,是一种动态的多层面翻译后修饰,并参与几乎所有的细胞过程,如细胞周期进程、信号转导、转录调节等[20]。1977年,Goldknopf等[21]首次发现Ub可通过赖氨酸连接修饰组蛋白。在20世纪80年代,研究表明三磷酸腺苷底物的泛素化与26S蛋白酶体降解相关联[22]。随后的研究发现,超过1 000种蛋白质调节人类细胞的泛素化,数千个蛋白质上存在数万个泛素化位点,大多数蛋白质在细胞生命周期的某个时刻都会经历泛素化,泛素化反应几乎涉及真核生物的所有代谢过程[22]。
研究表明,泛素化途径是在泛素-蛋白酶体系统(ubiquitin proteasome system,UPS)的催化下,泛素分子结合底物分子的过程[23]。该过程是由E1泛素活化酶、E2泛素缀合酶和E3泛素-蛋白质连接酶的作用介导[20,24],首先由E1激活泛素,然后E2将泛素转移到E3,最后E3催化泛素与靶蛋白的共价结合。相反,去泛素化酶(deubiquitinase,DUB)催化可介导催化底物蛋白中泛素分子的去除[24]。其中,E3决定泛素和底物连接的特异性和精确性,人类基因组里有超过800个E3,因此泛素化比其他蛋白质修饰更加复杂,功能更加多样[23]。
此外,泛素分子本身能以不同的结构形式结合效应蛋白,不同的效应蛋白可能具有不同的泛素结合域,效应蛋白以不同结合域特异性结合泛素,从而导致不同的功能,包括降解、信号转导和亚细胞定位的改变等。例如,K48(第48号赖氨酸)和K11链是蛋白酶体降解的关键信号,而K6、K27、K33和K63支链泛素链通常是不可降解的[24]。最近的研究[24-25]进一步揭示了支链泛素链在亚细胞定位或信号转导中的作用。
既往研究多聚焦于泛素化参与细胞凋亡、自噬等生理病理过程[26](图1),而近期研究表明,泛素化在CVD的发生发展中也具有调控作用。现就其作用进行总结和分析。
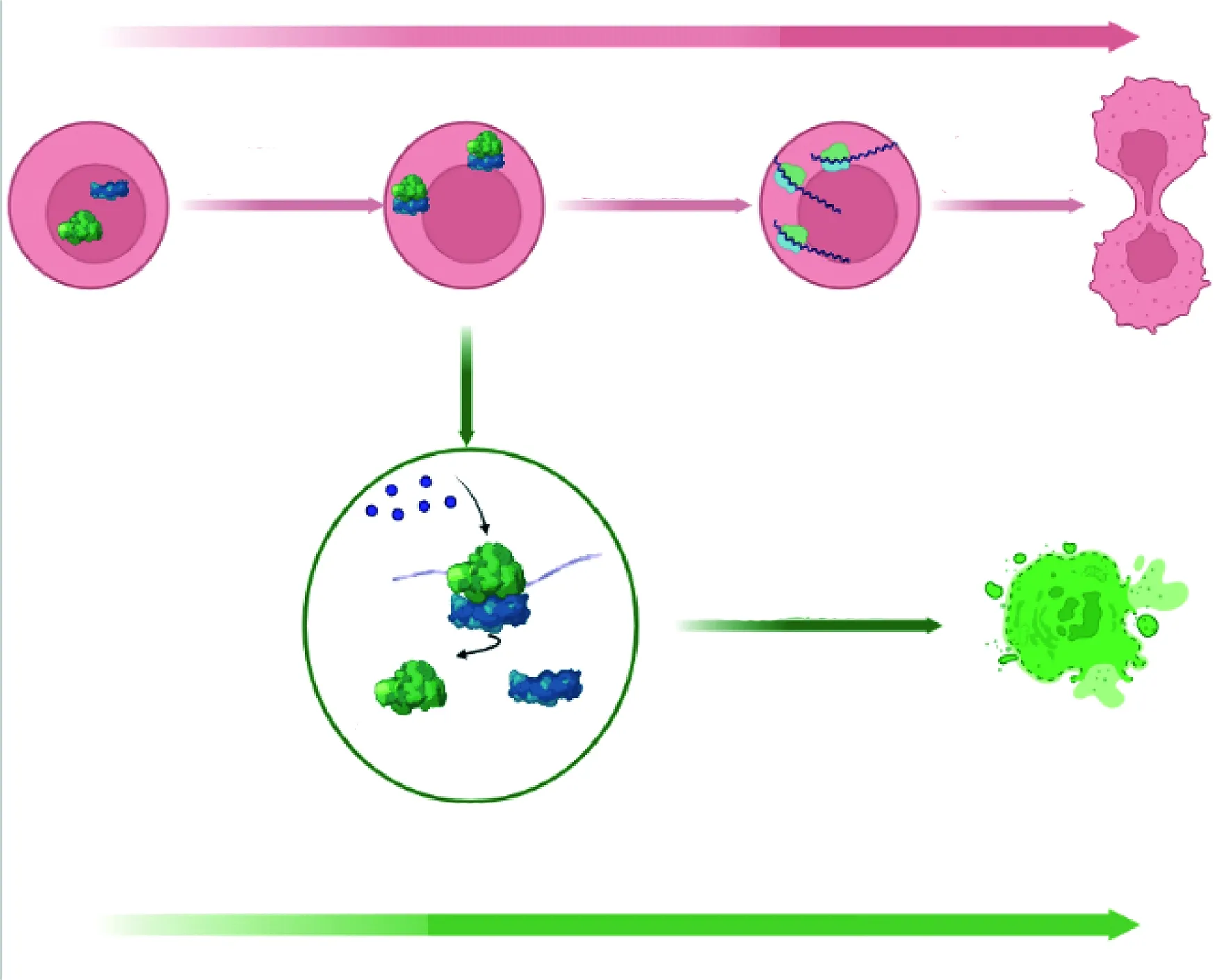
图1 泛素化与细胞损伤
2.1 核仁素泛素化与CVD
核仁素 (nucleolin,Ncl) 是核仁中最丰富的蛋白之一,主要分布于核仁区,也存在于核质、细胞质和细胞膜中,在核糖体新生中发挥重要作用[27]。自1973年Orrick等首次对其进行描述以来一直是众多研究的重点[27]。
Ncl主要由三个结构域组成:N端结构域、中心结构域和C端结构域。这三个结构域允许Ncl与不同的蛋白质和RNA序列相互作用,在核糖体新生的不同阶段发挥不同的作用[27]。在前核糖体RNA (pre-ribosomal RNA,pre-rRNA) 转录过程中,Ncl作为伴侣帮助pre-rRNA正确折叠、RNA包装、前信使RNA (pre-messenger RNA,pre-mRNA) 剪接和mRNA稳定[27]。Ncl还被证明参与细胞质分裂、细胞增殖、凋亡调节和应激反应等多个生理病理过程,从而参与血管生成、肿瘤发生、病毒感染等多个病理过程[27]。
近期研究表明,核仁素的泛素化可导致疾病的发生。2018年,Wang等[28]发现Ncl的泛素化会抑制Ncl蛋白表达,影响Ncl蛋白的稳定性,从而导致糖尿病肾病。糖尿病肾病是糖尿病常见的临床并发症,与CVD密切相关,是导致终末期肾脏疾病的主要原因。CYP4B1-PS1-001是长链非编码RNA,调节糖尿病肾病的纤维化[28]。Wang等[28]的研究发现在环己亚胺处理的系膜细胞中,CYP4B1-PS1-001的过表达导致Ncl泛素化,抑制Ncl蛋白表达,但不影响mRNA表达,这表明CYP4B1-PS1-001是通过泛素化修饰降解Ncl蛋白质,导致肾病[28]。另外,三重基序蛋白2 (tripartite motif-containing protein 2,TRIM2) 是Ncl泛素化中主要的E3泛素-蛋白质连接酶,敲除TRIM2基因会提高Ncl的蛋白水平,证明TRIM2作为E3泛素-蛋白质连接酶在Ncl降解中的作用。综上,研究提示核仁素的泛素化有望成为糖尿病肾病与CVD的治疗靶点。
此外,Ko等[29]发现多形性胶质母细胞瘤发生时,胶质瘤干细胞样细胞(glioma stem-like cells,GSC)的富集会上调鼠双微基因2(murine double minute 2,MDM2)介导的Ncl泛素化,从而降低Ncl水平。但抑制Ncl的去乙酰化会阻止Ncl泛素化的发生,STAT3和JNK信号通路也会阻止Ncl降解,从而阻止GSC的富集[29]。该发现表明Ncl有望成为多形性胶质母细胞瘤治疗的潜在靶点[29]。
2.2 核糖体泛素化与心肌重构
心肌肥厚和纤维化都会导致心肌重构,且都是心力衰竭发生发展的基础[30]。聚腺苷二磷酸核糖聚合酶[poly(ADP-ribose)-polymerase,PARP]1是PARP家族中的一员。PARP家族酶是CVD的重要损伤因子,尤其是在心脏瓣膜病中PARP起到重要作用。PARP主要负责将腺苷二磷酸核糖基团转移到靶蛋白质上,从而影响各种细胞过程[30-33]。PARP1激活后会识别受损的DNA片段,使片段发生多聚ADP核糖基化反应。 PARP1活性增强后,会与WW域E3泛素-蛋白质连接酶2(WW domain-containing E3 ubiquitin protein ligase 2,WWP2)作用并通过泛素化途径使得心肌细胞极度消耗能量,从而引发心肌细胞损伤,介导心肌重构[32]。这些研究表明PARP1泛素化程度影响心肌重构。
WWP2是与E6-AP羧基末端同源(homologous to the E6-AP carboxyl terminus,HECT)结构域家族类的E3泛素连接酶,在心脏中高表达,且参与异丙肾上腺素(isoproterenol,ISO) 诱导的心肌重构[30,34]。2020年,Zhang等[30]在小鼠心肌中特异性敲除WWP2,发现PARP1的表达增加但泛素化水平降低,多聚核糖基化水平升高,小鼠ISO诱导的心肌肥厚、纤维化和心力衰竭加重。当重新诱导WWP2的再表达会显著增加PARP1的泛素化,降低PARP1和多聚核糖基化水平,初步表明WWP2是一个调节ISO诱导心肌重构的保护因子[30]。Zhang等[30]进一步发现,在生理状态下,WWP2特异性结合到PARP1的乳腺癌易感基因羧基端结构域(the carboxyl-terminal domain of the breast cancer gene 1,BRCT),通过泛素化修饰PARP1的K249和K418位点促进PARP1降解,保护小鼠免受ISO诱导的心肌重构。研究表明WWP2在ISO诱导的心肌重构中起到重要作用,这为治疗与心脏重构相关的心脏病提供了新靶点[30]。
此外,在许多CVD中都观察到PARP1表达升高,PARP1活性升高,即多聚核糖基化现象[30]。PARP1和多聚核糖基化是重要的损伤因子,参与心肌细胞损伤和心肌重构的过程[30]。ISO诱导的心肌重构和心肌缺血再灌注损伤等条件下,心肌细胞产生活性氧和活性氮。这些活性物质引起DNA氧化损伤,进而激活PARP1产生多聚核糖基化反应,使心肌烟酰胺腺嘌呤二核苷酸和三磷酸腺苷水平显著下调,导致心肌细胞坏死[33]。综上,泛素化在心肌重构中起着举足轻重的作用。
2.3 核糖体泛素化与心肌梗死、缺血再灌注损伤
研究显示,在人类和各种心脏疾病的动物模型中(包括心肌梗死、缺血性心脏病和心力衰竭),Ub和活化的UPS的数量都在增加,泛素化相关基因在左心室坏死过程中上调,会激活UPS,激活的UPS导致细胞凋亡和心肌细胞收缩蛋白的丢失,导致左心室功能恶化[35]。
在压力过载小鼠模型中,蛋白酶体抑制剂Epoxomicin完全阻止了心肌肥厚,同时阻断了蛋白酶体激活,提示UPS可能参与心力衰竭的发生[35]。与核糖体60S亚基相关的E3泛素连接酶Listerin1(Ltn1),被证明可促进Nonstop降解和No-go降解途径产生的核糖体新生肽的降解,且依赖Ltn1的新生蛋白泛素化又导致核糖体亚基分解[36]。Rqc2是Ltn1招募到60S核糖体进行泛素化所必需的因子,与Cdc48和Npl4/Ufd1联合作用,促进核糖体进行泛素化降解[37]。
此外,Meyer等[38]通过多聚核糖体图谱发现,多聚体中存在G3BP1、G3BP2和USP10蛋白。通过对G3BP1和G3BP2双基因敲除(KO)的细胞培养,发现蛋白翻译会停滞,多聚核糖体减少为核糖体单体,并产生大量游离的核糖体60S亚基。对USP10-KO细胞进行泛素残留免疫亲和分析,发现泛素化的RPS2、RPS3和RPS10丰度最高,且在USP10-KO和G3BP1/2-双基因KO细胞中观察到40S和60S亚基中RPs的特异性降低。该研究还通过诱导核糖体停滞,发现去泛素化酶ZNF598介导的核糖体碰撞感应是导致培养细胞中RPS2、RPS3和RPS10单泛素化的主要原因,从而表明RPS2、RPS3和RPS10去泛素化作用于G3BP1-family-USP10复合物。USP10或G3BP1家族蛋白的敲除又增加了核糖体的降解和核糖体亚基的紊乱。
研究发现,泛素化酶MDM2,能使凋亡转录因子p53泛素化,且心肌细胞泛素化水平升高的同时,20S和26S活性降低[39]。在犬心肌缺血再灌注的模型中发现26S蛋白酶体活性和泛素化蛋白积累的下降[39],但在小鼠慢性心肌梗死模型中观察到蛋白酶体活性增加,包括UPS组分水平和蛋白酶体活性的增加,其在压力负荷诱导的小鼠心脏肥厚模型中也得到证实[35,39]。这些研究提示,泛素化在心肌梗死与心肌缺血再灌注损伤中的精准机制需深入研究,核糖体泛素化在其中的作用及机制亟待阐明。
2.4 核糖体泛素化与血管形成及相关疾病
RPs与血管形成密切相关[9]。在核糖体新生过程中,RPL5和RPL11作为复合体组装,被招募到60S核糖体亚基。游离核糖体的RPL5和RPL11与泛素化酶MDM2结合,抑制p53发生泛素化降解,导致p53转录活性增强、p53依赖的血管细胞周期阻滞[9]。在血管形成的过程中,游离的RPL17作为血管平滑肌细胞生长抑制剂,在小鼠中限制颈动脉内膜增厚。RPL17可抑制血管平滑肌细胞的生长,使其停留在G0/G1期。已知RPL23可通过抑制MDM2的功能来调控p53,以应对核糖体新生障碍[40]。RPL23的下调会诱导核糖体新生障碍,导致p53的稳定和激活,这表明RPL23在这一通路中既起效应又起传感作用[9]。因此,RPL17也可能是通过激活p53来调控细胞周期,尽管目前还没有证据表明其在p53通路中的作用[9]。
希佩尔-林道(von Hippel-Lindau,VHL)综合征是一种罕见的遗传性多器官肿瘤疾病,通过VHL突变基因传播,该疾病与嗜铬细胞瘤有关[41]。嗜铬细胞瘤是肾上腺髓质嗜铬细胞的非恶性肿瘤,会合成和释放大量儿茶酚胺并产生心血管病变。VHL综合征肿瘤抑制蛋白相关复合体(VHL tumor suppressor protein,pVHL)是缺氧诱导转录因子(hypoxia inducible factor,HIF)-1α亚基的E3泛素-蛋白质连接酶复合物的组成部分,能使HIF进行泛素化降解[42]。VHL综合征中pVHL功能的丧失导致HIF的异常积累,引起HIF下游基因的过表达,包括血管内皮生长因子的累积,这反过来又促进了血管性肿瘤的形成[42]。此外,核糖体中RNA聚合酶Ⅱ第七亚基(hsRPB7)是VHL泛素化的下游靶点,VHL可通过其β结构域直接与hsRPB7结合,靶向hsRPB7泛素化降解并降低血管内皮生长因子的表达来调控血管生成[41]。综上,核糖体泛素化在血管形成及相关疾病中的作用不容忽视。
3 展望
综上所述,RPs不仅是组成核糖体的重要部分,更是参与到各类生化反应的重要成分,具有多种核糖体外功能,如RPs泛素化具有重要的生理病理作用。近年来关于RPs泛素化在疾病发生方面的研究主要聚焦肿瘤的发生,少有在CVD方面的研究,且具体作用机制尚未阐明。实验大多局限于体外细胞模型,缺少动物实验与临床研究,在动物实验基础上推导的结论及假说也亟需验证。本文概述核糖体泛素化在CVD中的作用,旨在引起研究者关注,并在此领域进一步探索,为治疗CVD提供新策略。
作者贡献声明李玉洁:综述构思及撰写,图表绘制;张瑜:综述构思及修订;吴诚洁,陆琳,王燕丽:综述修订;李杨欣:综述构思,图表设计及修订;李玉洁,张瑜:并列第一作者
利益冲突所有作者均声明不存在利益冲突
猜你喜欢
内蒙古民族大学学报(自然科学版)(2022年2期)2022-11-22 06:44:43
世界最新医学信息文摘(2020年68期)2020-12-25 11:55:27
肿瘤防治研究(2020年5期)2020-07-09 13:06:08
生物学教学(2019年9期)2019-09-23 03:53:02
吉林大学学报(医学版)(2015年4期)2015-12-17 07:48:13
浙江大学学报(农业与生命科学版)(2015年4期)2015-12-15 12:47:42
中国医学科学院学报(2015年5期)2015-03-01 04:03:46
现代检验医学杂志(2015年2期)2015-02-06 02:01:01
四川生理科学杂志(2014年3期)2014-02-28 14:09:38
中国医学科学院学报(2013年6期)2013-03-11 20:26:01
