
猪伪狂犬病毒gE蛋白单克隆抗体的制备及竞争ELISA抗体检测方法的建立
2022-11-04 07:37:02杨世丽周海欧李翠婷马燕梅
江西农业学报 2022年8期
袁 梦,常 巍,杨世丽,周海欧,李翠婷,马燕梅
(福建农林大学 动物科学学院(蜂学学院),福建 福州 350002)
伪狂犬病(Pseudorabies,PR)严重威胁着猪养殖业,其病原体伪狂犬病毒(Pseudorabies virus,PRV)是一种囊膜病毒,其遗传物质是线性双链DNA分子,编码至少70种蛋白质[1-2],可感染多种动物。研究表明PRV在小鼠体内可引起细胞因子风暴以及细胞炎症性坏死[3]。猪是PRV的唯一储存宿主,母猪感染PRV后易出现不孕、流产,仔猪感染PRV后会出现腹泻、呼吸衰竭甚至死亡[4]。成年猪感染PRV康复后经常形成潜伏感染,带毒的猪不发病,但在一定条件下会释放病毒,成为猪群中的主要传染源[5]。有研究表明,PRV在中国猪群中依然存在持续传播,并且存在跨物种传播现象[6]。有报道指出PRV可以感染人,导致人眼内炎和急性脑炎,并在患者脑脊液中分离出了PRV毒株hSD-1/2019[7]。因此如何准确、便捷地检测PRV已成为亟待解决的问题。gE蛋白包含577个氨基酸[8],是由us8基因编码形成异二聚体的囊膜糖蛋白[9];gE是PRV主要的囊膜毒力蛋白[10],编码囊膜蛋白gE的基因变异程度低,该蛋白能介导PRV与宿主动物细胞的融合,促进PRV的扩散与释放,导致PRV对宿主体内嗜神经组织的趋向性,有利于PRV在高级神经中枢神经突触间传递扩散[11-12]。用缺失gE基因的PRV毒株感染动物,病毒在动物鼻腔上皮细胞中的复制不受影响,但其在神经组织中产生的伤害显著下降[13]。目前生产上常见的PRV基因缺失株疫苗多缺失gE基因,因此检测gE可以区分野毒株和疫苗株[9]。重组PRV gE可用作血清诊断试验,检测其特异性抗体并区分是否为疫苗接种动物[14],因此制定针对PRV gE抗体的检测方法具有十分重要的意义。
本试验先对gE蛋白进行原核表达并纯化,在免疫BALB/c小鼠后制备gE单克隆抗体,建立竞争ELISA抗体检测方法,制备抗体检测试剂盒,为获得一种更廉价有效的检测方法寻找新途径。
1 材料与方法
1.1 试验材料
PRV FA株和SP2/0细胞由本实验室保存;DH5α和BL21(DE3)感受态细胞购自福州晴百旺生物科技有限公司;EasyPure Quick Gel Extraction Kit、EasyPure Viral DNA/RNA Kit、完全弗氏佐剂和不完全弗氏佐剂购自全式金生物技术有限公司;无内毒素质粒中提试剂盒购自康为世纪生物科技股份有限公司;DNA纯化试剂盒购自威格拉斯生物技术有限公司。
1.2 质粒构建以及动物免疫
1.2.1gE基因的扩增及质粒构建 从NCBI上下载PRV FA株的全基因序列(KM189913),用Snapgene软件根据PRV gE基因主要抗原表位区段(561 bp)设计1对引物:gE-F 5′-CGGGATCCGAGGCC GACGACG-ATGACCTCAA-3′(BamH I);gE-R 5′-CGGAATTCCGAGAAGAGCTGCGAGTGGAA -3′(EcoR I)。提取PRV-FA株的DNA为模板,以94 ℃ 5 min;94 ℃ 30 s,65 ℃ 30 s,72 ℃ 1 min,35个循环;72 ℃ 5 min的条件进行PCR扩增,对产物进行1%琼脂糖凝胶电泳,在核酸凝胶成像仪的紫外灯下将目的片段切下。
1.2.2 pET-28a-gE表达质粒的构建 将目的片段与载体分别用限制性内切酶BamH I和EcoR I进行双酶切,载体酶切体系:载体pET-28a 2 μL、BamH I 1 μL、EcoR I 1 μL、10×Buffer 4 μL、ddH2O 32 μL。目的片段酶切体系:目的片段30 μL、BamH I 1 μL、EcoR I 1 μL、10×Buffer 4 μL、ddH2O 4 μL。将经金属浴(37 ℃ 1 h)双酶切后的载体与目的片段用DNA纯化试剂盒纯化,然后用T4 DNA连接酶进行连接,连接体系:目的片段 8 μL、载体DNA 2 μL、T4 DNA连 接 酶1 μL、10×Buffer 1.5 μL、ddH2O 2.5 μL。将连接体系在金属浴(16 ℃)中连接过夜。用连接产物转化DH5α感受态细胞,涂具有卡那抗性的LB固体培养基平板,第2天挑取菌落至具有卡那抗性的LB液体培养基中,在摇床(37 ℃、200 r/min)上培养12 h,再用无内毒素质粒中提试剂盒提取质粒,进行双酶切鉴定。将鉴定正确的质粒送北京擎科生物技术有限公司进行测序。
1.2.3 重组蛋白gE-6×His的诱导表达和可溶性鉴定 将测序正确的质粒转化BL21(DE3)感受态细胞,涂具有卡那抗性的LB固体培养基平板,第2天挑取菌落至具有卡那抗性的LB液体培养基中,在37 ℃、200 r/min的摇床上培养4 h;用1.2节中的方法,将菌液作为模板,进行菌液PCR鉴定,将阳性菌按菌液∶培养基=1∶100的体积比加入具有卡那抗性的LB液体培养基中,置于37 ℃、200 r/min的摇床上培养1~2 h;当菌液的OD600值约为0.6时,加入终浓度为0.5 mmol/L的IPTG,在25 ℃、200 r/min的摇床上培养6 h,然后在4 ℃下以8000 r/min的转速离心5 min,收集菌体沉淀,进行超声波破碎,将破碎后的菌体沉淀进行SDS-PAGE电泳,电泳凝胶用考马斯亮蓝染色液染色后用清水脱色,观察目的蛋白条带。
1.2.4 重组蛋白gE-6×His的纯化 使用结合缓冲液平衡镍柱,将蛋白样本通过恒流泵流入镍柱;再用结合缓冲液洗涤镍柱,依次用100、200、300、400和500 mmol/L咪唑浓度的洗脱缓冲液洗脱目的蛋白,收集从镍柱底端流出的各浓度的洗脱缓冲液,进行SDS-PAGE电泳,电泳凝胶用考马斯亮蓝染色液染色后用清水脱色,观察蛋白的纯化情况。
1.2.5 小鼠免疫 将纯化后的蛋白与佐剂按体积比1∶1乳化,然后免疫BALB/c小鼠,采用背部皮下多点注射的方法进行免疫,每只小鼠每次免疫注射100 ng重组蛋白gE-6×His,免疫间隔14 d。首次免疫采用完全弗氏佐剂,二免和三免均采用不完全弗氏佐剂,三免后对小鼠进行尾部采血,检测小鼠血清抗体效价。选择血清效价最高的小鼠,在三免后第14天不用任何佐剂,将100 ng纯化后的融合蛋白注射小鼠腹腔,进行加强免疫。
1.3 竞争抑制ELISA检测方法的建立
1.3.1 最佳反应条件的确定 用方阵滴定法确定抗原浓度及免疫鼠血清稀释度,将融合蛋白进行倍比稀释,包被96孔酶标板,将纯化后的蛋白用包被缓冲液倍比稀释到浓度分别为2500、1250、625、312.5、156.25、78.15、39.06、19.53、9.77、4.88和2.44 ng/mL,按100 μL/孔加入酶标板。一抗使用的免疫鼠血清的倍比稀释度为从1∶1000到1∶64000,每个蛋白包被的浓度设1个PBS阴性对照。二抗使用PBS配制的3%脱脂牛奶的羊抗鼠HRP,稀释度为1∶2000。显色后加终止液,测定OD450值,OD450值≈1的孔对应的抗原包被浓度和一抗稀释度为最佳条件。
1.3.2 血清的最佳稀释度 根据优化后的竞争ELISA条件,分别取阳性和阴性待检血清,按1∶2、1∶4、1∶8和1∶16进行倍比稀释,用1.3.1节中的方法进行检测,计算血清抑制率(%),确定待检血清的最佳稀释度。
1.3.3 阴阳性判断标准的确定 对24份PRV阴性血清用已建立的竞争ELISA方法进行检测,计算血清抑制率,并按公式“阴阳性临界值=X+3×SD”计算阴阳性临界值,其中X是检测的阴性血清抑制率的平均值,SD是其标准方差。
1.3.4 临床检测 使用建立的ELISA方法检测32份待测猪血清,同时运用PCR方法进行检测,并比较两者的检测结果。
1.4 单克隆抗体的制备
1.4.1 细胞融合 在小鼠加强免疫后第3天进行细胞融合,于细胞融合前1 d取非免疫BALB/c小鼠腹腔的巨噬细胞,铺96孔细胞培养板,制备饲养层细胞。在细胞融合前将小鼠脱颈处死,于75%乙醇中浸泡5 min,在超净台中无菌取小鼠脾脏,在200目细胞筛上将脾脏研碎,用在4 ℃下储存的无血清1640培养基冲洗细胞筛,将收集的滤液以2000 r/min离心10 min,弃上清液,加10 mL 1640培养基,并进行细胞计数。选状态良好的SP2/0细胞,按SP2/0细胞∶脾细胞=1∶5的比例混匀,以2000 r/min离心10 min;弃上清液,在1 min内加入40 ℃水浴的50% PEG 4000溶液,滴加同时混匀;在90 s内加入37 ℃水浴的无血清1640培养基,加至50 mL时终止融合,将混合细胞以2000 r/min离心10 min;弃上清,用37 ℃水浴的20% FBS HAT 1640选择培养基稀释细胞至每毫升2×106个脾细胞,加入96孔板(已有饲养层细胞),每孔加入100 μL,于37 ℃、5% CO2条件下培养。
1.4.2 杂交瘤细胞的筛选 于细胞融合后第4天,用20% FBS HAT 1640选择培养基半量换液;在细胞融合后第8、9和12天,用20% FBS HT 1640选择培养基半量换液。从第12天到第16天,用1.3.1节中建立的间接ELISA方法检测上清抗体,阴性、阳性对照分别为SP2/0上清和免疫鼠血清。用间接ELISA方法检测上清抗体应重复2~3次,对稳定分泌抗体的孔进行亚克隆。用有限稀释法将阳性细胞进行连续2~3次的亚克隆,用间接ELISA方法检测上清抗体,当用于筛选的孔中的上清液阳性率达到100%时,即表明筛选到可以稳定产生单一抗体的杂交瘤细胞。
1.4.3 小鼠单抗腹水诱导 提前1 d向10周龄BALB/c小鼠腹腔注射不完全弗氏佐剂0.5 mL。第2天将处于对数生长期且生长状态良好的杂交瘤细胞吹下,以800 r/min离心5 min,用PBS洗涤3次,进行细胞计数;加适量PBS,调整细胞密度至每毫升2×106个杂交瘤细胞;向小鼠腹腔注射该液0.5 mL,在注射后7~10 d小鼠腹腔膨大,用注射器收集腹水,以8000 r/min离心10 min,收集上清液,保存于-80 ℃。
1.5 单克隆抗体的鉴定
1.5.1 单克隆抗体效价的测定 将腹水上清从1∶1000开始稀释,测定OD450nm值。选择OD450nm值≥0.2、P/N值≥2.1且最高稀释倍数为最佳稀释度效价。
1.5.2 单克隆抗体的Western-blotting鉴定 提取攻毒PRV的PK15细胞蛋白样本,将未攻毒的细胞样作为阴性对照,使用Western-blotting鉴定免疫原性。
1.5.3 单克隆抗体的特异性检测 使用间接ELISA方法检测单克隆抗体的特异性,以PRV、PRRSV、VSV包被酶标板,杂交瘤细胞上清为一抗,羊抗鼠HRP为二抗检测抗体的特异性。
1.6 数据处理与分析
使用SPSS软件对试验数据进行t检验、成对双样本均值分析,与对照组相比,**、****、NS分别表示P<0.01、P<0.0001、不显著。
2 结果与分析
2.1 质粒构建及蛋白纯化
2.1.1gE基因的扩增及pET-28a-gE表达质粒的构建 以PRV PK-15培养物为模板,用设计的特异性引物进行扩增,并通过1%琼脂糖凝胶电泳进行检测。在图1A中,扩增片段PRV gE主要抗原表位区段约为573 bp,符合预期;用内切酶BamH I和EcoR I酶切重组质粒,证明pET-28a-gE构建完成。图1B是pET-28a-gE的酶切结果,得到预期大小的条带(目的片段大小约为573 bp)。质粒送擎科公司测序,结果正确。

图1 gE基因的扩增及pET-28a-gE表达质粒的构建
2.1.2 重组蛋白gE-6×His的诱导表达和可溶性鉴定 pET-28a-gE表达融合蛋白gE-6×His是在25 ℃、0.5 mmol/L IPTG、150 r/min的条件下诱导6 h后获得的。与pET-28a-gE诱导前的表达产物比较,pET-28a-gE诱导后出现了1条25~35 kDa的条带,大小约为25.7 kDa(图2A),符合预期,说明融合蛋白gE-6×His诱导表达成功。从图2B可以明显看出融合蛋白gE-6×His在上清中。

图2 重组蛋白gE-6×His的诱导表达和可溶性鉴定结果
2.1.3 重组蛋白gE-6×His的纯化 融合蛋白诱导后超声破碎,进行gE-6×His和gH-6×His镍亲和层析,图3是可溶性融合蛋白gE-6×His的纯化结果,可见纯化后条带单一,可用作抗原进行免疫。

图3 融合蛋白gE-6×His的纯化结果
2.2 竞争抑制ELISA检测方法的建立
2.2.1 棋盘滴定法确定最佳反应条件 通过方阵滴定试验,以PRV做抗原包被,以gE单克隆抗体做检测抗体,结果显示,当PRV包被的稀释度为1.25 μL/mL、单克隆抗体的稀释度为1∶400时,OD450值为0.95(表1)。

表1 竞争ELISA检测方法的优化结果(OD450值)
2.2.2 待检血清最佳稀释度的确定 通过方阵滴定试验,以PRV做抗原包被,以gE单克隆抗体做检测抗体,试验结果如表2所示,当血清的稀释度为1∶2时,阳性、阴性的抑制率均最高,分别为49%和5%。
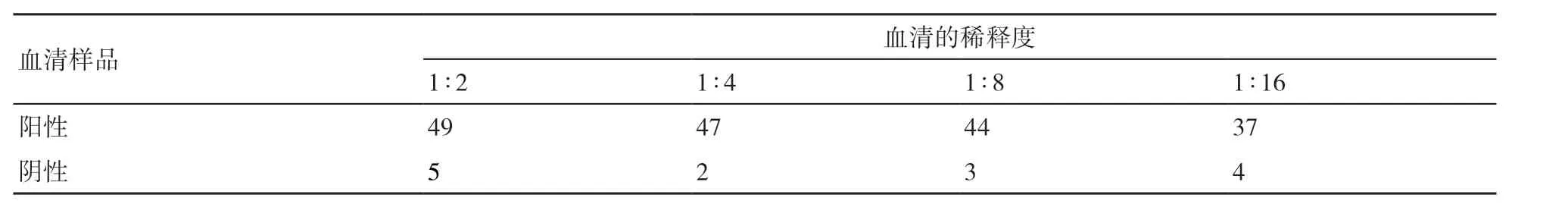
表2 血清最佳稀释度的确定结果(抑制率) %
2.2.3 阴阳性判断标准的确定 用以上建立的竞争ELISA方法检测24份gE多抗阴性血清,其抑制率平均值(X)为9.98,标准方差(SD)为2.83,根据公式:阴阳性临界值=X+3×SD,得出gE多抗血清的阴阳性临界值为18.47。
2.2.4 临床检测 用gE单克隆抗体建立的竞争ELISA抗体检测方法检测32份血清,检出7份阳性、25份阴性(表3),与PCR检测结果(图4)相符。

表3 临床检测结果

图4 32份血清的PCR检测结果
2.3 单克隆抗体的制备
2.3.1 间接ELISA检测鼠血清抗体方法的优化 用方阵滴定法确定抗原包被的浓度和抗体的稀释度。当融合蛋白gE-6×His包被的浓度为0.63 μg/mL、阳性血清的稀释度为1∶128000时,OD450值为1.01(表4)。

表4 间接ELISA检测鼠gE抗体方法的优化结果(OD450值)
2.3.2 间接ELISA检测杂交瘤细胞株 通过间接ELISA方法检测杂交瘤细胞株上清的抗体效价,得到了7株细胞,将其分别命名为1C1、1B2、1D5、3D6、3G6、3E7、3C10。
2.3.3 阳性杂交瘤细胞的亚克隆 细胞融合后的阳性孔中通常有不止1种杂交瘤细胞,为了得到单克隆杂交瘤细胞,用有限稀释法筛选单个杂交瘤细胞,结果得到2株能够稳定产生抗体的杂交瘤细胞1B2和3E7(图5)。

图5 杂交瘤细胞的形态
2.4 单克隆抗体的鉴定
2.4.1 单克隆抗体效价的测定 取过度培养的杂交瘤细胞上清和小鼠腹水,从1∶10开始稀释,用间接ELISA方法检测效价,结果细胞上清的抗体效价为1∶2560,小鼠腹水的抗体效价为1∶5120(表5)。

表5 单克隆细胞培养基抗体效价的检测结果(OD450值)
2.4.2 杂交瘤细胞稳定性的鉴定 杂交瘤细胞冻存可能会影响其产生抗体的能力,为了确定其能否稳定产生抗体,用间接ELISA方法对复苏后的细胞培养上清进行检测。结果发现冻存前和复苏后产生抗体的能力变化小,单克隆细胞3E7的抗体分泌能力更稳定(图6)。
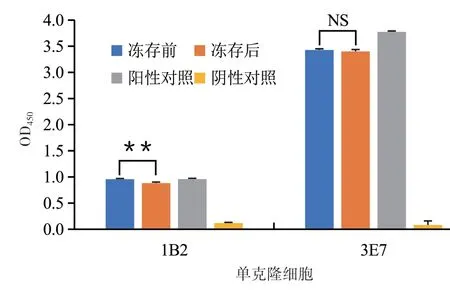
图6 杂交瘤细胞稳定性的检测结果
2.4.3 单克隆抗体的特异性检测 用间接ELISA方法检测单克隆抗体的特异性,以PRV、PRRSV、VSV包被酶标板,以杂交瘤细胞上清为一抗,结果表明,单克隆抗体具有特异性(图7)。

图7 间接ELISA方法检测单克隆抗体的特异性
2.4.4 单克隆抗体的Western-blotting鉴定 将PK15细胞作为阴性对照,用Western-blotting鉴定单克隆抗体的特异性,结果显示PK15 PRV-FA培养物出现了1条大小约120 kDa的单一条带,而阴性对照没有条带,说明单克隆抗体能特异性地与PRV结合,特异性高(图8)。

图8 单克隆抗体的Western-blotting鉴定结果
3 讨论
生猪集约化养殖现今已成为人类获取可食用猪肉的主要方式,猪皮也可制成皮革,具有很高的经济价值,猪产业已经成为经济发展和日常生活中不可或缺的一部分[15]。PRV在国内外都有报道,且有多种PRV疫苗用于预防该病毒[16]。在临床上区分野毒感染猪和疫苗免疫猪对于近年来的猪场PRV净化工作有重要意义[17]。目前有多种方法可检测PRV,有多重RT-PCR检测技术[18]、动物接种法、血清学实验等。由于进口的PRV抗体ELISA试剂盒价格高,难以应用于大面积检测,因此开发特异、敏感、简捷且价格低廉的PRV抗体检测试剂盒十分必要。
gE蛋白是PRV的囊膜蛋白,有暴露于表面的抗原位点,其中gE是PRV主要的囊膜毒力蛋白[19],可介导PRV与宿主细胞的融合。现有研究表明在PRV疫苗开发中,重组疫苗已成为新趋势[20-21]。由于该蛋白的基因高度保守,多数PRV缺失株缺失gE蛋白,于是本研究选取了含有gE 5个主要抗原表位区段的基因(561 bp),通过PCR的方法将PRV FA株的gE 5个主要抗原表位区段成功扩增并构建至pET-28a(+)载体上,转化原核表达菌株BL21(DE3),通过原核表达系统成功表达了融合蛋白gE-6×His。
经镍柱亲和层析后获得了纯化的融合蛋白gE-6×His;将gE-6×His与免疫佐剂乳化后免疫BALB/c小鼠,建立了间接ELISA方法,以检测免疫后血清效价,融合后也用此方法筛选杂交瘤细胞。将不完全弗氏佐剂注入小鼠腹腔,1 d后向腹腔中注射杂交瘤细胞悬液,约7 d后取小鼠腹水。本试验用的小鼠腹水诱导方法更快捷,且不完全弗氏佐剂更容易引起特异性免疫应答[22],注射后1 d即可注射杂交瘤细胞,缩短了腹水诱导的时间,与杨国平等[23-24]使用的降植烷和石蜡相比更加快速。用Western-blotting鉴定单克隆抗体,可见在约120 kDa处有1个明显条带,与吴桐[25]制备的gE单克隆抗体的条带位置相近,但本研究制备的单克隆抗体Western-boltting条带更单一,特异性更强。
本研究采用PRV-FA作为抗原包被酶标板,以gE单克隆抗体作为检测抗体,建立了PRV的竞争性ELISA检测方法,并优化了检测条件,这为开发特异、敏感、简捷且更廉价的PRV检测试剂盒奠定了基础,对当下伪狂犬病的净化工作有一定的指导意义。
猜你喜欢
中国现代医药杂志(2020年10期)2020-12-14 07:20:14
食品科学(2018年10期)2018-05-23 01:27:28
现代检验医学杂志(2016年3期)2016-11-15 01:59:28
畜牧兽医学报(2016年5期)2016-07-16 06:09:08
西南医科大学学报(2015年1期)2015-08-22 13:01:46
医学研究杂志(2015年3期)2015-06-10 06:41:52
医学信息(2015年14期)2015-04-29 11:58:11
特产研究(2015年1期)2015-04-12 06:36:20
中国当代医药(2015年9期)2015-03-01 02:01:59
西南军医(2015年6期)2015-01-23 01:25:50
