
线粒体相关内质网膜在脑卒中后认知障碍中作用的研究进展*
2022-11-01 03:25:34陈淑颖白艳杰陈丽敏丁志敏张雍闯李晓晓
中国病理生理杂志 2022年10期
陈淑颖, 白艳杰, 陈丽敏, 丁志敏, 王 岩, 张雍闯, 李晓晓
(1河南中医药大学康复医学院,河南 郑州 450046;2河南中医药大学第一附属医院康复中心,河南 郑州 450099)
脑卒中后认知障碍(post-stroke cognitive impairment,PSCI)常出现记忆、学习等多个认知域的损伤,大约有1/3 的脑卒中患者会出现不同程度的认知障碍,对康复进程和生活质量均造成较大影响[1-2]。脑卒中后的继发性改变十分复杂,研究表明,脑卒中缺血缺氧造成的炎症反应、钙紊乱、线粒体功能障碍等病理过程的激活是启动细胞凋亡、自噬等死亡机制,加重脑神经元损伤,导致不同程度认知障碍的重要因素[3]。其中,线粒体外膜在细胞凋亡中有关键作用:活化的Bax、Bcl-2 拮抗因子等促凋亡蛋白能聚集在线粒体外膜上改变其通透性,并促进线粒体细胞色素C(cytochrome C,CytC)等凋亡诱导物的释放[4]。此外,研究表明,线粒体相关内质网膜(mitochondriaassociated endoplasmic reticulum membrane,MAM)在维持细胞稳态中具有重要作用,线粒体外膜和内质网之间通过Ca2+、需肌醇酶1α(inositol-requiring enzyme 1α,IRE1α)、磷酸弗林蛋白酶酸性氨基酸簇分选蛋白2(phosphofurin acidic cluster sorting protein 2,PACS2)等参与细胞凋亡信号传导,进而影响细胞的命运[5]。下调 MAM 相关蛋白 PACS2 可使海马神经元和星形胶质细胞变性而出现认知障碍,并且在痴呆患者脑组织中观察到MAM介质表达减少[6]。同时出于脑组织的高能量需求,神经元常因为线粒体功能障碍而加重损伤,由于MAM 支持线粒体质量控制,因而MAM 还可能通过控制线粒体质量影响神经功能[7]。因此,调控MAM 或许是避免脑卒中后病理过程加剧,保护神经元免受损伤的有效途径。现主要针对MAM在PSCI中的具体作用展开综述,以期为临床靶向治疗提供参考资料。
1 线粒体与内质网相互作用构成MAM
MAM亦称线粒体-内质网结构偶联,是线粒体和内质网的直接接触位点,是线粒体外膜与内质网在物理结构和生物化学上相互通信的一个结构域。在物理结构方面,MAM 通过连接物决定其接触位点的横向范围和间隙距离,其中蛋白的种类可以限制线粒体与内质网的接触范围,因此在不同组织中,接触区的范围是变化的[8]。MAM 可以招募多种蛋白参与维持其基本结构,包括线粒体融合蛋白、肌醇1,4,5-三磷酸受体(inositol 1,4,5-triphosphate receptors,IP3Rs)、电压依赖性阴离子通道(voltage-dependent anion channels,VDAC)、葡萄糖调节蛋白75(glucose-regulated protein 75,Grp75)、线粒体分裂蛋白1(mitochondrial fission protein 1,Fis1)、B 细胞受体相关蛋白31(B-cell receptor-associated protein 31,BAP31)、蛋白酪氨酸磷酸酶相互作用蛋白51(protein tyrosine phosphatase-interacting protein 51,PTPIP51)和囊泡相关膜蛋白相关蛋白B(vesicle-associated membrane proteinassociated protein B,VAPB)等[9]。MAM 通过招募不同的膜蛋白及调控不同的信号传导来维持线粒体及内质网等细胞器的正常功能,从而影响细胞的命运。
在生物功能方面,线粒体与内质网之间的串扰主要与各种细胞信号传导有关,包括脂质生物合成和转运、钙信号转导、线粒体动力学等,通过接触点的信号变化影响MAM 的功能[10]。线粒体与内质网之间的连接物是MAM 发挥各种生物功能的基本支架,这些膜蛋白决定了线粒体和内质网的动态结构及功能。如MAM 招募线粒体融合蛋白2(mitofusin 2,Mfn2)、发动蛋白相关蛋白1(dynamin-related protein 1,Drp1)等调节因子,通过内质网膜收缩线粒体分裂位点等物理操作调节线粒体分裂/融合以维持正常的线粒体形态结构[11]。哺乳动物细胞在MAM还具有复杂的蛋白复合物,在控制细胞稳态平衡中具有重要作用。其中Mfn2-Mfn1/2、IP3Rs-Grp75-VDAC、VAPB-PTPIP51 及 Fis1-BAP31 等蛋白复合物相互作用,促进或抑制MAM 的连接,介导MAM 间的通信,它们对MAM 结构的复杂调节作用在各种神经系统疾病中有重要影响[5]。
2 MAM参与PSCI病理进程的发生发展
2.1 调控脂质代谢 在真核细胞中,磷脂主要在线粒体内膜和内质网中合成,维持膜正常的脂质代谢合成与转运对维持细胞正常的活性至关重要,而MAM就是促进脂质正常转运的一个关键平台[12-13]。大脑是脂质含量最丰富的器官之一,研究表明随着年龄的增长,磷脂酰肌醇、磷脂酰乙醇胺和磷脂酰胆碱在大脑中的水平随着年龄的增长而缓慢下降,对痴呆患者大脑进行检查时显示病变区域脂质提取物中的磷脂酰肌醇、磷脂酰乙醇胺和磷脂酰胆碱含量较高或仅有轻度减少[14]。李昊[15]在检验患者血清时,观察到PSCI患者血清总胆固醇和低密度脂蛋白胆固醇水平均较高,证实高脂是PSCI的独立危险因素,这进一步证实了脂质代谢异常极有可能是PSCI的发病机制之一。在给年幼和老年大鼠连续喂食高脂肪/高胆固醇饮食后,不仅显示出海马tau蛋白磷酸化异常,其空间工作记忆也均出现较差的表现[16]。而临床应用抗脂质代谢异常药物能够减轻认知障碍,如黄腐酚及其两种氢化衍生物α,β-二氢黄腐酚和四氢黄腐酚治疗就可改善高脂诱导的空间学习和记忆缺陷[17]。MAM富含几种脂质转移蛋白和生物合成酶,包括胆固醇酰基转移酶/甾醇O-酰基转移酶1、二酰基甘油O-酰基转移酶2等,若MAM功能障碍,很容易导致脂质代谢异常;在痴呆患者脑组织中就曾检测到脂质异常并伴有MAM 锚定蛋白胆固醇酰基转移酶/甾醇O-酰基转移酶1 含量及活性的上调[18-19]。由此可见,脂质代谢异常能够诱发PSCI,而MAM 介导的脂质合成和转运在维持细胞正常生命活动中发挥重要作用,因此MAM功能异常有可能是PSCI的发病机制之一。
2.2 调控钙平衡 细胞器之间常通过Ca2+信号交流,而钙平衡对于维持细胞正常的能量代谢及生命活动至关重要[20]。MAM 中存在高钙域,其通过IP3Rs-Grp75-VDAC 复合物和 sigma-1 受体(sigma-1 receptor,Sig-1R)等调控着钙释放和钙转运,线粒体将它们解码为特定的信号以调节细胞新陈代谢和凋亡等基本功能[21]。Chalmers等[22]发现,在脑部缺血损伤6 h 后小鼠脑组织中钙/钙调蛋白依赖性蛋白激酶T286 残基的磷酸化增加,相反抑制钙/钙调蛋白依赖性蛋白激酶的自主活性则可以防止海马缺血性损伤,对海马具有神经保护作用。此外,一项随机对照实验显示PSCI 模型大鼠Ca2+浓度升高,电针可以通过下调Ca2+浓度,使脑梗死体积减少及空间学习和记忆能力增强[23]。这也进一步证实了上述观点,钙超载诱发的脑神经元损伤可能是导致PSCI 的重要因素。Sig-1R 是MAM 的重要组成蛋白,并在线粒体应激条件下与IP3Rs 结合,从而促进Ca2+转移并调节钙稳态[5]。Li 等[24]通过免疫荧光实验检测显示,使用Sig-1R激活剂SA4503可显著增加海马神经元的轴突长度,对神经功能障碍产生重大影响,并且SA4503的生长促进作用伴随着对电压门控钙离子通道的抑制,这有力地证明了MAM 组成蛋白通过调控钙转运参与海马神经元的轴突生长。同时有研究表明,在痴呆患者体内Sig-1R 通路被破坏,恢复Sig-1R 稳态则可以发挥神经保护作用,减轻认知障碍[25],这与上述激活Sig-1R 能增加海马神经元轴突长度影响神经元功能的结果一致。此外,钙紊乱还对细胞凋亡具有重要影响。缺血性卒中的发作会导致神经元产生过高的线粒体活性氧以及Ca2+超载引起兴奋性氨基酸毒性,进而诱导线粒体通透性转换孔开放,最终将驱动线粒体凋亡诱导蛋白CytC排入细胞质并触发细胞凋亡[26]。MAM 锚定蛋白VDAC在影响细胞凋亡中发挥重要作用。在脑缺血期间VDAC 可促进线粒体释放CytC到细胞质基质而诱导细胞凋亡,同时VDAC又是调节Ca2+运输到线粒体的关键组件,其与IP3Rs和Grp75 共定位在MAM 上,增强线粒体对钙的吸收[27-28]。因此,VDAC除了会直接诱导细胞凋亡外,还有可能通过钙转运间接诱导神经元凋亡而加重神经缺损。以上证据提示,MAM 功能蛋白异常可能会影响海马神经元轴突生长并通过影响钙转运诱导凋亡,加重海马神经元损伤,而适当的调控则有可能促进缺血后神经元的恢复并减轻认知障碍。
2.3 调控线粒体动力学 线粒体动力学包括融合和分裂两个过程,这对维持线粒体稳态平衡和细胞存活具有重要作用。因此,线粒体动力学失衡常伴随活性氧异常升高、钙超载和能量缺陷等线粒体功能受损现象。线粒体融合与分裂失衡是导致卒中后钙超载、氧化应激和细胞凋亡等病理过程进而引起神经元损伤及认知障碍的重要因素[29]。研究表明,在缺血性体内及体外模型中均存在Mfn2 的减少,这造成线粒体融合异常并进一步导致细胞凋亡,而诱导Mfn2 上调则可以恢复线粒体功能、发挥抗凋亡作用以减少海马神经元的损伤[30]。海马神经元除了线粒体融合障碍之外,线粒体分裂也存在异常,在痴呆和轻度认知障碍患者的外周血淋巴细胞中均显示Drp1 蛋白显著降低而 Fis1 蛋白表达增加[31]。Jiang等[32]通过敲除小鼠海马和大脑皮层神经元中的Mfn2从反面验证了线粒体功能形态变化及其继发氧化应激反应、炎症反应造成海马神经元损伤并参与认知障碍的发生发展。这提示线粒体融合-分裂失衡与其继发病理改变共同作用,共同影响着PSCI 的病理进程,调控线粒体动力学有可能是治疗PSCI 的一个新靶点。此外,Mfn2 大量富集于MAM 上,它不但促进线粒体外膜的融合,还参与维持MAM 的基本结构及功能,MAM 的蛋白复合物Fis1-BAP31 也参与构建用于凋亡信号传导的支架[7,33]。欧阳梦琪等[34]发现,脑卒中模型大鼠海马CA1区MAM结构疏松,且Mfn2蛋白减少,而辣椒素通过抑制MAM 结构疏松、上调Mfn2 使大鼠学习记忆能力明显改善。由此可知,MAM 也是调节线粒体动力学的重要结构,且MAM调控的线粒体动力学影响PSCI 的转归及预后,这为治疗PSCI提供了一个可能的新靶点。
2.4 调控内质网应激(endoplasmic reticulum stress,ERS) 当内质网受到缺血缺氧等外界刺激时,内质网内蛋白稳态失衡,即发生ERS,此时内质网会通过未折叠蛋白反应调整内部程序,以恢复稳态;若不能恢复,则调整到凋亡信号[35]。MAM 已被证明通过蛋白激酶R 样内质网激酶(protein kinase R-like endoplasmic reticulum kinase,PERK)和 IRE1α 等折叠酶参与未折叠蛋白反应启动程序以维持细胞正常功能[36]。除此之外,MAM 还可以通过 ERS 促进炎症小体的激活,ERS 与炎症反应及细胞凋亡之间相互影响。一项体外细胞培养实验表明,过表达IRE1α 可增强ERS 和神经元凋亡[37]。此外,一项动物实验表明内源性磷酸化IRE1α 蛋白表达增加可以导致核苷酸结合寡聚化结构域样受体蛋白3(nucleotide-binding oligomerization domain-like receptor protein 3,NLRP3)炎症小体活化及明显的神经行为缺陷,在给予IRE1α抑制剂后的进一步研究证实ERS 可能通过加剧炎症反应参与 PSCI 的发生[38]。有趣的是,NLRP3 炎症小体的激活反过来也会诱导和加重ERS,它们之间存在着复杂的相互作用[39]。由此可见,ERS与炎症反应共同作用加剧神经元损伤,有效调控ERS 对预防和治疗PSCI 具有重要作用。最新研究表明,PERK 和IRE1α 等酶富集在 MAM 中,PERK 除了参与未折叠蛋白反应外,对维持MAM 结构和功能的完整性也至关重要[40]。MAM介导的ERS还可以影响脑卒中后神经元凋亡。Fei 等[41]的研究表明,激活 PERK 可加重ERS和凋亡活性,而抑制PERK信号通路则能减少细胞凋亡,并减轻缺血再灌注损伤并减轻认知障碍。基于这一点,Li等[42]发现,敲除ERS 调节相关因子发状分裂相关增强子,促进了ERS 诱导的细胞凋亡,而应用PERK 抑制剂则显著减弱了ERS 诱导的中风后神经元凋亡及认知障碍恶化。这进一步证实,ERS除了与炎症反应共同作用外,还直接诱导神经元凋亡,从而造成PSCI。因此,有理由相信MAM 相关蛋白介导的ERS 和其继发炎症反应及细胞凋亡在脑卒中后神经元损伤造成的认知障碍中具有重要影响。
2.5 调控线粒体自噬 线粒体自噬能够清除损伤或老化的线粒体,维持细胞稳态,对细胞命运具有重要影响。其中线粒体自噬在PSCI 中的作用具有双重性,适度自噬能清除堆积的异常物质而起到神经保护作用,过度自噬则会导致不受控制的降解而诱导更多的神经元死亡[43]。一项动物实验表明,脑缺血再灌注损伤大鼠存在自噬标志物LC3-II 和含FUN14 域 蛋 白 1(FUN14 domain containing 1,FUNDC1)表达升高[44]。并且抑制自噬或可减轻PSCI[45]。但是 Wang 等[46]的研究表明,电针灸减轻缺血再灌注后神经元损伤的机制在于促进PTEN 诱导激酶1(PTEN-induced kinase 1,Pink1)/parkin 介导的线粒体自噬。这也再次印证了线粒体自噬作用的两面性及其在PSCI 发生发展中的重要作用。此外,介导线粒体自噬的Pink1 及FUNDC1 蛋白均参与维持MAM 的完整性,MAM 是自噬体的起源部位,同时VAPB-PTPIP51 复合物也参与调节自噬[47]。VAPBPTPIP51 是MAM 重要的蛋白复合物,参与维持MAM的基本结构及功能。VAPB-PTPIP51 对线粒体自噬起负性调节作用,而阻断IP3Rs 介导的Ca2+运输则能影响这种作用[48]。痴呆患者颞叶神经元自噬激活而VAPB 和 PTPIP51 却降低,并且 VAPB 和 PTPIP51 的丧失会进一步破坏突触活性影响神经突触功能[49],这也再次印证了上述观点。MAM 在其中发挥重要作用:一方面MAM 的功能蛋白直接介导线粒体自噬影响神经功能;另一方面MAM 的信号传导功能如钙转运异常可能也会影响自噬。虽然目前线粒体自噬作为靶点可以治疗PSCI,但对其研究仍然浅显,如何适度调控线粒体自噬将其积极效应最大化或许是未来研究的热点。因此,是否可以通过MAM 调节线粒体自噬影响PSCI的进程尚需深一步探究。
MAM在PSCI中作用的总结如图1所示。
3 小结与展望
综上所述,现有证据表明MAM 不同程度地参与了PSCI的病理进程,其各项生理功能与PSCI病理机制存在诸多交织。一方面MAM 本身异常会导致钙转运和脂质代谢等细胞信号传导失灵;另一方面它还会引起线粒体和内质网功能障碍,进而影响细胞生存,加重机体病理损伤及不良事件的发生。MAM结构及功能正常才能维持细胞正常运转,若MAM 异常,不但会直接诱导细胞凋亡,还会进一步加重氧化应激和炎症反应等继发改变,导致更多神经元死亡,加重认知障碍。
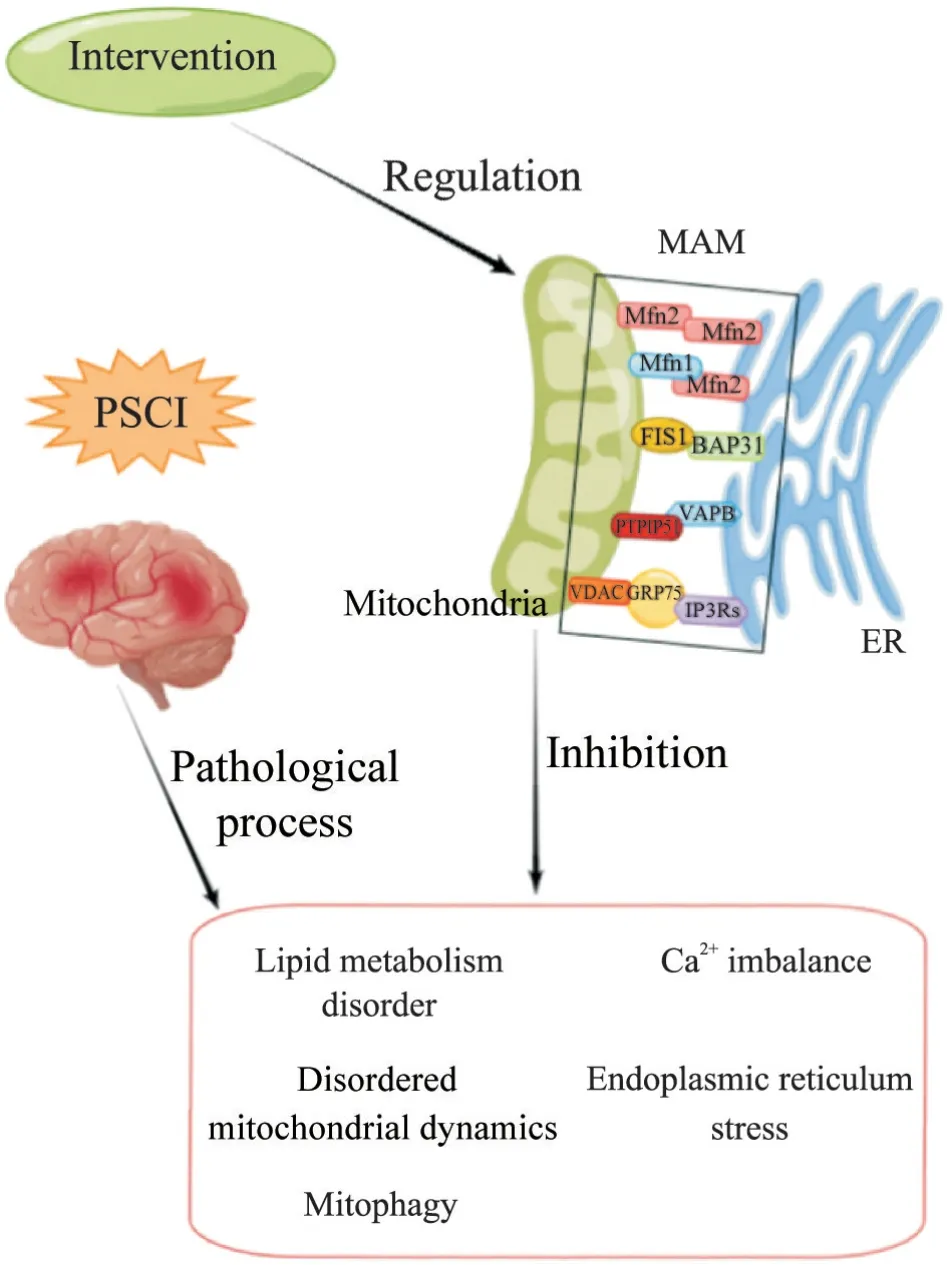
Figure 1. Potential targets of mitochondria-associated endoplasmic reticulum membrane(MAM)in post-stroke cognitive impairment(PSCI). When PSCI occurs,it is often accompanied by pathological changes such as lipid metabolism disorder,calcium imbalance,mitochondrial dynamic imbalance,endoplasmic reticulum stress,and mitophagy. MAM also participates in the pathological process of PSCI through related physiological functions.Therefore,targeting and regulating related pathological mechanisms through MAM may alleviate the occurrence and development of PSCI. ER:endoplasmic reticulum;Mfn2:mitofusin 2;Mfn1:mitofusin 1;FIS1:fission 1 homologue;BAP31:B-cell receptor-associated protein 31;PTPIP51:protein tyrosine phosphatase-interacting protein 51;VAPB:vesicle-associated membrane protein-associated protein B;VDAC:voltage-dependent anion channels;GRP75:glucose-regulated protein 75;IP3Rs:inositol 1,4,5-triphosphate receptors.图1 MAM在PSCI中的潜在作用靶点
虽然目前通过MAM 改善PSCI 相关的实验研究较少,但毋庸置疑的是MAM在PSCI各种病理生理改变中具有重要作用。通过调控MAM 之间的串扰及各种功能蛋白或许能够影响脑卒中的病理改变,并进一步减轻PSCI 的发生发展。然而究竟是脑卒中引起MAM 功能障碍使继发病理改变加重导致认知障碍还是PSCI进一步引起MAM异常,又或是二者彼此影响,尚需深一步研究。鉴于MAM 结构复杂及功能蛋白多样,未来还需深入研究在相应的病理过程中MAM的变化机制并进一步明确MAM在PSCI病理过程中的可作用靶点,以期特异性识别MAM,精准调控影响细胞命运的信号传导,为今后PSCI 的靶向药物开发提供新的参考资料。
猜你喜欢
保健医苑(2022年5期)2022-06-10 07:47:36
解放军医学杂志(2021年12期)2022-01-18 03:53:24
中老年保健(2021年6期)2021-08-24 06:54:06
现代临床医学(2021年1期)2021-01-26 00:55:52
医学新知(2019年4期)2020-01-02 11:03:54
中成药(2018年9期)2018-10-09 07:18:36
中成药(2018年1期)2018-02-02 07:19:53
中成药(2017年4期)2017-05-17 06:09:26
安徽医科大学学报(2016年12期)2017-01-15 14:21:55
中国当代医药(2015年33期)2015-03-01 02:09:08
