
线粒体相关内质网膜调节神经元自噬的研究进展*
2022-11-01 03:25:34王一聃丁旭东陈俊文
中国病理生理杂志 2022年10期
王一聃, 曾 炼, 丁旭东, 桑 明, 陈俊文
(湖北医药学院附属襄阳市第一人民医院呼吸与危重症医学科,湖北 襄阳 441000)
神经元作为不可分裂的细胞,相比其他细胞寿命更长,在发育与维持过程中,需去除错误折叠的蛋白以及损伤衰老的细胞器,以保证生命活动的正常进行[1]。细胞自噬是细胞清除代谢物的主要途径,其目的在于保证细胞对能量的需求以及内环境的稳定。自噬是维持正常神经功能所必须的,参与神经元的轴突运输,突触可塑性以及包括线粒体在内的细胞器的质量控制等过程[2-3]。当神经元自噬障碍时,随着蛋白质与损伤细胞器的积累,会诱发神经退行性变[4];当神经元自噬过度激活时,也会继发神经元的坏死,提示适度的自噬水平是神经元发挥正常功能的基础。然而,目前对于神经元自噬发生的分子机制尚不清楚。越来越多的证据表明线粒体相关内质网膜(mitochondria-associated endoplasmic reticulum membranes,MAMs)在其中发挥重要作用。作为线粒体与内质网之间信号与物质交换的桥梁,MAMs具有广泛的生物学功能,参与了Ca2+信号转导,脂质代谢,线粒体动力学调控以及细胞自噬等[5]。通过对MAMs 中的蛋白构成进行蛋白质组学分析,检测到大量调控细胞自噬的蛋白质(表1)[6-7]。同时MAMs 结构与功能的异常被证实参与神经退行性疾病的发病过程,而目前对于MAMs 在神经元自噬中的作用尚不清楚,本文汇总了MAMs 参与调节神经元自噬和神经退行性疾病的研究进展。
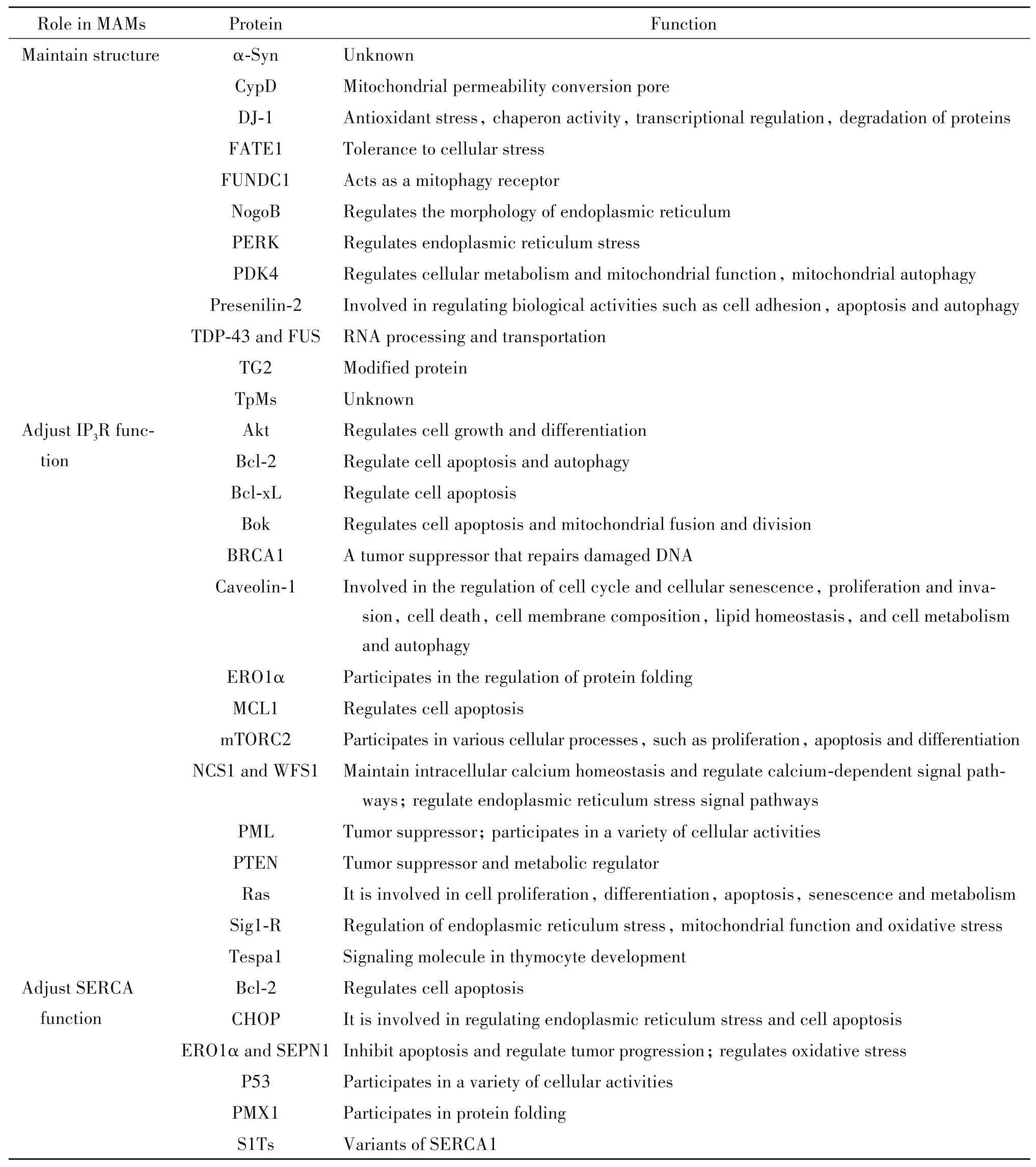
表1 MAMs中维持结构及发挥调节功能的蛋白[7]Table 1. Proteins in MAMs that maintaining structure and regulating functions
1 MAMs的构成与功能
真核细胞具有多种功能各异的膜结构细胞器,它们之间存在信号与物质的交换,而这种交换依赖于细胞器之间的特殊连接,其中MAMs 是典型的位于线粒体与内质网之间接触的膜结构。MAMs 于上个世纪50 年代首次被观察到,后续的研究表明大约5%~20%的线粒体外膜与内质网相互关联[8]。从结构上来看,MAMs的厚度即线粒体外膜与内质网间的距离约为10~25 nm,其中粗面内质网与线粒体间的MAMs 厚度更大[9]。除厚度外,MAMs 形态也各具差异,根据内质网小管包绕线粒体的比例将MAMs 分为3型,其中1型包绕比例约占10%,2型则近乎完全包绕,3型仅包绕50%的线粒体,大多数MAMs属于1型[10]。从功能上来看,MAMs早期主要作为脂质合成的场所,其中首次检测到的蛋白是参与脂质代谢的蛋白质,包括磷脂酰乙醇胺甲基转移酶2(phosphatidylethanolamineN-methyltransferase 2,PEMT2)、磷脂酰丝氨酸合酶 1/2(phosphatidylserine synthase 1,PSS1/2)等,而这些蛋白也成为鉴定MAMs 可靠的标志性蛋白[11]。除此之外,MAMs主要的功能是参与调节钙稳态,进一步调控三磷酸腺苷(adenosine triphosphate,ATP)合成,线粒体分裂和细胞死亡等生命活动。而这种作用主要依赖于IP3Rs/GRP75/VDAC1 信号途径实现[5]。作为包绕线粒体的膜结构,MAMs 参与调节线粒体的运动与分裂。线粒体具有沿微管运动的特性,受Ca2+调节,故MAMs 通过调控钙转运来影响线粒体运动[12]。同时,MAMs 也调控线粒体分裂。其中发动蛋白相关蛋白1(dynamin-related protein,DRP1)由胞质转运至线粒体作为驱动线粒体分裂的关键环节[13]。研究证实 DRP1 在 MAMs 中表达,同时还包括两种驱动DRP1线粒体转运的其他蛋白,分别是线粒体分裂因子(mitochondrial fission factor)和分裂蛋白 1(fission protein 1)[14],因此 MAMs 可通过调节它们的表达与转运进而调控线粒体分裂。
2 神经元自噬的概述
细胞自噬从形成活化的Unc-51样自噬激活激酶1 复合体开始,在级联反应作用下,激活beclin-1,参与合成Ⅲ类磷脂酰肌醇3-激酶(phosphatidylinositol 3-kinase,PI3K)复合体,驱动自体吞噬。形成的自噬小体经由自噬相关蛋白ATG12-ATG5 以及LC3-Ⅱ(ATG8-Ⅱ)构成的复合体调控,进而驱动自噬体的成熟[15]。成熟的自噬体与溶酶体连接,降解细胞中多余的成分。依据识别的特异性,自噬被分为选择性自噬和非选择性自噬。其中非选择性自噬以饥饿诱导的自噬为代表,目的在于细胞组分的再利用[16],而选择性自噬具有一个或多个受体介导的识别过程,该受体通过与LC3 相互识别将待降解物与自噬体连接,促进自噬体包绕[15]。依据自噬发生部位的不同,神经元自噬分为轴突自噬,树突自噬和胞体自噬[3,17]。其中轴突中的自噬较为多见,神经元自噬体由轴突中产生后往胞体运输,而这种运输是在驱动蛋白作用下沿微管双向进行的,最终在胞体中代谢清除[18]。除轴突自噬外,树突中也检测到了细胞自噬,其作用主要是调节神经元的兴奋性,当使用低剂量N-亚硝基二甲胺刺激原代大鼠海马神经元后,可观察到树突棘中自噬体数量的增多[19]。相比轴突和树突,胞体中的自噬相对较少,且自噬体的成熟程度也较低[17]。胞体自噬主要目的是去除损伤及衰老的线粒体即线粒体自噬(mitophagy),进一步维持神经元线粒体结构与功能的完整[20]。依据发生时间的不同,神经元自噬又可分为发育期的自噬和成熟期的自噬。自噬是神经元发育所必须的,抑制胚胎期神经元的自噬可以导致皮层中神经元数量的减少[21]。同时自噬还可以调节神经元轴突生长,以及突触的形成等发育过程[22-23]。对于成熟的神经元,自噬的主要作用是维持神经元的内稳态,而自噬功能异常则是大多数神经退行性疾病发生的重要基础[24]。
3 MAMs调节神经元自噬
MAMs 对于神经元自噬的调节作用主要体现在以下几方面:(1)对来源神经元的MAMs 中所含蛋白质进行蛋白质组学分析,检测到大量与自噬相关或调节自噬的蛋白[6-7]。其中包括线粒体融合蛋白2(mitofusin 2,MFN2),MFN2 表达上调时,可削弱神经元中线粒体与内质网间的相互作用,干扰线粒体的正常功能,并促进MAMs 中的钙转运,转运至线粒体的Ca2+通过激活DRP1 促进线粒体分裂以及PTEN诱导的假定激酶1(PTEN-induced putative kinase 1,PINK1)介导线粒体自噬,从而诱发神经退行性疾病[25]。除MFN2 外,磷酸呋喃酯酸性簇分选蛋白2(phosphofurin acidic cluster sorting protein-2,PACS-2)也被证实表达于MAMs 中,其功能主要是参与调节MAMs 的结构和细胞自噬。当下调神经元中PACS-2的表达时,除了影响MAMs 结构的完整外,还会导致脂质合成功能障碍,从而阻止LC3 与磷脂酰乙醇胺(phosphatidylethanolamine,PE)的偶联抑制细胞自噬[26]。(2)MAMs参与自噬体的形成,被认为是神经元自噬的潜在起源。自噬体的发生受自噬相关蛋白的调控,神经元MAMs 中被证实含有大量自噬相关蛋白,包括ATG14 和ATG5 等,当自噬被激活时,MAMs中表达的ATG14L,ATG5 和含双FYVE 结构的蛋白1(double FYVE-containing protein 1,DFCP1)的含量增多,在它们协同作用下,驱动细胞自噬[27]。相反的,当抑制MAMs 中的蛋白表达时,可以检测到神经元中自噬体数量的减少[28]。(3)MAMs 通过干预脂质合成调控神经元自噬,LC3 与PE 偶联被认为是自噬体成熟的标志,而PE 主要是通过内质网合成后转运至MAMs 进行加工分泌,在此过程中受MAMs 的调控,故MAMs 可以通过影响PE 的合成来参与调控神经元自噬[29]。(4)MAMs 通过调节 Ca2+转运来调控神经元自噬,Ca2+作为调控神经元生命活动的重要信号分子,也参与了介导神经元自噬的发生尤其是线粒体自噬,当神经元线粒体功能障碍时,由内质网经MAMs 转运至线粒体的Ca2+诱发线粒体钙超载,导致线粒体膜电位的降低,进一步激活线粒体自噬。而当破坏MAMs 介导的Ca2+转运时,会导致AMPK 易位至MAMs 中进一步诱导神经元发生自噬性坏死[30]。总之,MAMs对于神经元自噬的调节主要基于其物质与功能的共同作用。
4 MAMs调控神经元线粒体自噬
线粒体自噬是损伤线粒体通过选择性自噬途径转运至溶酶体中进行代谢清除的过程,其目的在于维持线粒体质量的稳定。线粒体自噬需要受体介导,其中主要的信号途径即PINK1/parkin 途径,当线粒体损伤时,聚积于线粒体中的PINK1 招募parkin并磷酸化激活它,parkin 活化后泛素化修饰p62/SQSTM1等受体蛋白,介导线粒体自噬[31]。线粒体自噬对于维持神经元正常的生理功能具有重要意义,同时也参与了众多神经病理性过程。无论是脑损伤还是神经退行性疾病,线粒体自噬功能障碍被认为是疾病发生的重要基础[32-33],尤其在神经退行性疾病中,由于神经元中积累着大量错误折叠的蛋白质,他们一方面抑制线粒体自噬来逃脱降解,另一方面过度激活线粒体自噬损害功能正常的线粒体,最终造成神经元的死亡[33]。MAMs作为线粒体外膜的延续,也参与调控神经元线粒体自噬。其中PINK1 被证实在神经元MAMs中有表达,同时兼具募集parkin的功能,当线粒体功能障碍时,储存于MAMs 中的PINK1可以转运至线粒体中招募并激活parkin,驱动线粒体自噬[34]。除 PINK1/parkin 途径外,MAMs 还可通过影响含FUN14 结构域蛋白1(FUN14 domain-containing 1,FUNDC1)的表达调控线粒体自噬。FUNDC1主要参与缺氧条件下诱导的线粒体自噬,与PINK1/parkin途径不同的是,FUNDC1 可以直接与LC3 结合,但这个过程受到氧气的调控[35]。除直接激活FUNDC1外,MAMs 通过促进 FUNDC1 与 IP3Rs 的相互作用来进一步增加MAMs 中Ca2+转运影响线粒体自噬[14]。除了调节 Ca2+转运外,MAMs 中的 FUNDC1 对 MAMs本身也有作用,当抑制FUNDC1 表达时,会导致MAMs 的蛋白丰度减少包括参与调节细胞自噬的组分,从而干扰其对于神经元自噬的调控[36]。
5 MAMs相关的神经元自噬与神经退行性疾病
MAMs 结构与功能的异常被证实参与神经退行性疾病的发病过程。MAMs 主要通过调控β-淀粉样蛋白(amyloid β-protein,Aβ)和α-突触核蛋白(αsynuclein,α-Syn)等错误折叠蛋白合成、神经元钙稳态、神经元线粒体功能、脂质代谢、神经元自噬等参与神经退行性疾病的发生过程[37]。由于MAMs 中富含乙酰辅酶A 乙酰转移酶1(acetyl-CoA acetyltransferase 1,ACAT1)和胆固醇,两者都是调节自噬的重要分子,因此MAMs 被认为可通过增强细胞自噬,调控阿尔茨海默病(Alzheimer disease,AD)患者和动物模型中 Aβ 和 tau 蛋白的清除[38]。此外,神经元线粒体功能障碍是AD 发生的重要基础,其通过增加MAMs 介导的活性氧的合成,诱导内质网应激,进而减少自噬体形成,促进tau蛋白的沉积[39]。除AD 外,MAMs 中还存在大量调控线粒体自噬的相关蛋白包括PINK1/parkin,其中parkin 的突变被证实是常染色体阴性遗传帕金森病患者发病的重要原因,其表达异常会诱导线粒体肿胀,引起线粒体损伤,最终导致黑质-纹状体中多巴胺能神经元进行性减少并伴随着 α-Syn 的沉积[28]。由 MAMs 蛋白功能障碍导致内质网-线粒体接触失调通过影响神经元自噬活性从而介导肌萎缩侧索硬化症(amyotrophic lateral sclerosis,ALS)的发生,其中多个参与调节ALS 的关键基因 包 括SQSTM1、OPTN、UBQLN2和VCP等 都 在MAMs 中有所表达[40]。总之,MAMs 调控神经元自噬进而调节神经退行性疾病依赖于其内容物成分的变化,主要体现在调节自噬相关蛋白的表达发生改变。
6 总结与展望
自噬维持神经元内稳态,同时也是神经元发育和发挥神经功能的基础。MAMs 作为内质网与线粒体间的桥梁,通过多种途径介导神经元自噬(图1):一方面,它充当了自噬相关蛋白执行功能的平台,参与自噬体的形成与成熟;另一方面通过调控Ca2+转运,脂质代谢影响自噬体的结构与功能。总的来说,MAMs 对于神经元自噬的调节是基于其物质与功能的联合作用。MAMs 被揭示参与神经退行性疾病的发生,同时大多数神经退行性疾病都涉及到神经元自噬功能的异常。虽然目前已有研究揭示了MAMs参与调控细胞自噬,甚至被认为是自噬发生的潜在起源,但仍有许多问题要我们进一步的探索。MAMs究竟是通过表达哪些蛋白参与调节神经元自噬体的形成与成熟?这个过程是否还受到其他因子的调控?它们是否与MAMs的其他功能包括Ca2+转运,脂质代谢之间存在相互关联?MAMs 结构与功能的异常,神经元自噬的异常,神经退行性疾病三者之间的联系?随着技术的不断发展,MAMs的成分和功能被深入挖掘,其在神经元自噬过程中的作用会被进一步阐明,有望成为治疗神经退行性疾病的有效靶点。
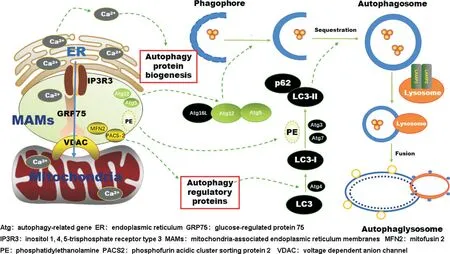
Figure 1. Schematic diagram of MAMs regulating neuronal autophagy. MAMs are involved in the genesis and maturation of autophagosomes by regulating calcium ion transduction,biosynthesis of autophagy-related proteins,and the expression of autophagyregulatory proteins.图1 MAMs调控神经元自噬示意图
猜你喜欢
解放军医学杂志(2021年12期)2022-01-18 03:53:24
海洋通报(2021年1期)2021-07-23 01:55:14
生物学通报(2021年4期)2021-03-16 05:41:26
现代临床医学(2021年1期)2021-01-26 00:55:52
老年医学与保健(2017年6期)2017-02-06 05:30:03
安徽医科大学学报(2016年12期)2017-01-15 14:21:55
中华肩肘外科电子杂志(2017年1期)2017-01-11 03:27:59
中外医疗(2015年5期)2016-01-04 03:57:57
中国当代医药(2015年33期)2015-03-01 02:09:08
癌变·畸变·突变(2014年1期)2014-03-01 04:39:36
