
分光光度法氨氮精密度偏性试验研究
2022-10-18 13:46:36高甲弟
地下水 2022年5期
高甲弟
(商洛水文水资源勘测中心,陕西 商洛 726000)
水环境监测分析测试机构实验室,为了确保监测数据的“五性”,即准确性、可靠性、真实性、完整性和可比性。针对所承担的监测项目逐步开展精密度偏性基础实验,进一步验证监测方法、监测人员的分析水平、分析仪器、化学试剂纯度、分析环境条件等影响因素是否受控;对该项目的空白试验、加标回收试验、国家标准物质测定的精密度、准确度、空白试验值等质量控制图的应用、测定时的影响因素及消除方法等做进一步试验研究与探讨。
1 基本概况
1.1 分析方法
HJ535-2009 水质氨氮的测定纳氏试剂分光光度法。
1.2 实验原理
在水中氨氮是以铵离子或游离态的氨等形式存在,遇到纳氏试剂会反应生成红棕色络合物,该络合物的吸光度与氨氮含量成正比,测定吸光度应选定波长420 nm处。
1.3 氨氮的测定范围
0.1~2.0 mg/L。对于超范围的样品可以适当稀释(浓缩),调整到曲线中间浓度,予以测定。
1.4 影响因素及消除方法
水样中含有有机物、钙离子、镁离子、悬浮物、硫化物和余氯时会对测定结果产生影响,若水样中存在以上物质时应进行合理的处理,测定结果才能真实、可靠。
若样品中存在余氯,可加入适量的硫代硫酸钠溶液去除,用淀粉-碘化钾试纸检验余氯 是否除尽。在显色时加入适量的酒石酸钾钠溶液,可消除钙镁等金属离子的干扰。若水样浑浊或有颜色时可用预蒸馏法或絮凝沉淀法处理。
1.5 仪器
50 ml比色管、7220分光光度计、20 mm比色皿、移液管、容量瓶、烧杯。
1.6 试剂纯度与材料
该实验所用试剂均为国药分析纯,应按规范要求准备实验用水,应使用经计量单位检定合格的量器和容量器皿。
1.6.1 无氨水
在无氨环境中制备,纯水器法。
1.6.2 主要试剂
纳氏试剂(HgI2-KI-NaOH)、酒石酸钾钠溶液(KNaC4H6O6·4H2O,500 g/L)、硫代硫酸钠溶液(Na2S2O3,3.5 g/L)、硫酸锌溶液(ZnSO4·7H2O,100 g/L)、氢氧化钠溶液(NaOH,250 g/L、1 mol/L)、盐酸溶液(HCl,1.18 g/mL、1 mol/L)、硼酸溶液(H3BO3,20 g/L)、淀粉-碘化钾试纸、氨氮标准贮备溶液(1 000 μg/mL)、氨氮标准工作溶液(10 μg/mL)。
1.7 人员
熟悉该国标分析方法,掌握溶液配制,了解分光光度计的使用与维护,经过岗位培训且持有国家认可委监发的《水环境监测从业证书》的分析员。
2 试验分析过程
根据《水环境监测规范》(SL219-2013)要求,须对空白、0.1 C、0.5 C、0.9 C、天然水样(自制水样)等五种样品进行连续六天的平行测定,以确定其方法空白、标准样质量是否符合规范要求,分析过程如下。
2.1 试剂配制
按照HJ535-2009 水质氨氮的测定纳氏试剂分光光度法中的试剂配制要求进行。
2.2 样品的配制
根据GB7479-1987 水质氨氮的测定纳氏试剂分光光度法所示,方法测定上限与校准曲线最高点浓度值相同,均是2.0 mg/L,即C=2.0 mg/L。在 50 ml比色管中加入氨氮标准工作溶液1 ml、5 ml、9 ml,分别加入超纯水至刻度线,充分混匀,配制成0.1 C、0.5 C、0.9 C、天然水样(自制水样)
2.3 标准曲线的绘制及合理性分析与样品分析
2.3.1 标准曲线的绘制与合理性分析
在 8个50 ml比色管中,依次加入氨氮标准工作溶液 0.00、0.50、1.00、2.00、4.00、6.00、8.00 和 10.00 ml ,对应的氨氮含量分别为 0.0、5.0、10.0、20.0、40.0、60.0、80.0 和 100 μg,稀释至50 ml。加入 1.0 ml酒石酸钾钠溶液,摇匀,再加入纳氏试剂 1.0 ml,摇匀。放置 10 min后,在波长420 nm下,用20 mm比色皿,以水作参比,测量吸光度。标准曲线、空白、0.1 C、0.5 C、0.9 C、天然水样(自制水样)、标准样品、加标样品连续六天,在同环境条件下,进行样品平行测定,并记录. 从表1来看,各曲线相关系数>0.999、各曲线截距<0.005、各曲线斜率均在误差允许范围内。各次工作曲线是合理的。
2.3.2 样品分析
按照标准曲线的测定方法与步骤,每天测得各样品数据、各样品的平均值、标准差分别按照公式(1)、(2)计算详见表2。
(1)
(2)

表1 曲线合理性分析表

表2 样品检测数据统计表
3 成果评价
3.1 估算分析方法的检测限
空白批内标准偏差按照式(3)计算
(3)
式中:Swb为空白批内标准偏差;n为每批测定个数;Xij为为各批所包含的各个测定值;i为代表批;j为代表同一批内各个测定值。
采用表2中空白样品的测定值按公式(3)计算Swb=0.003 7。
因空白测定次数少于20次,因此方法检出限(MDL)用公式(4)计算:
(4)
式中:MDL为方法检出限;tf为显著水平为0.05(单侧),自由度为f时的t值;f为批内自由度;m为批数;n为每批测定个数;Swb为空白平行测定。
按公式(4)计算MDL=0.021。
3.2 各样品精密度分析
各样品批内变异(a)按照公式(5)计算,各样品批间变异(b)按照公式(6)计算,并与F检验的临界值比较,F计算值小于F临界值为两组数据精密度无显著性差异;反之两组数据精密度存在显著差异。
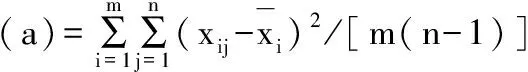
(5)
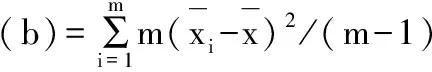
(6)
从表3可以看出,6月15日到6月20日各样品测定与平行测定,所获得的两组数据精密度无显著性差异。表明分析人员操作水平、仪器与环境、化学试剂等满足国标的要求,分析质量可靠。
3.3 回收率
根据回收率公式P(%)=(c2×V2-c1×V1)/(c0×V0)×100%
(7)
式中:c0为标准溶液浓度mg/L;c1为加标前溶液浓度mg/L;c2为加标后溶液浓度mg/L;v0为加标体积ml;V1为加标前溶液体积ml;V2为加标后溶液体积ml。

表3 各样品精密度分析表
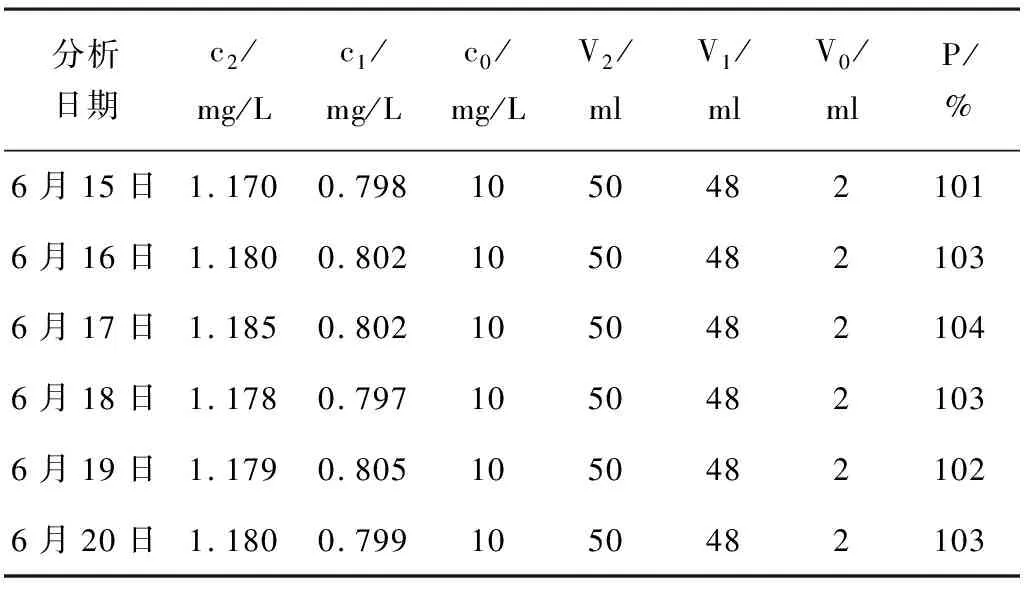
表4 各批次加标样回收率计算表
求得各批次的回收率分别按照公式(7)计算,分别101%、103%、104%、103%、102%、103%。
平均回收率按照算数平均法计算,平均回收率为103%。当回收率在95%~105%时合格,因为本次试验平均回收率为103%,平均回收率合格,表明实验室测定数据,准确度高,数据可靠(见表4)。
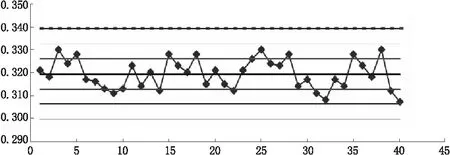
图1 统一样品(标样)质量控制图
3.4 合理性检查
通过绘制统一样品(标样)质量控制图见图1,将检测数据顺序点在图上:由图1实验数据都落入控制限内在标准值上下波动,则表明试验过程未失控;在中心线同一侧未出现连续7点,则表明测定数据正常;测定数据在标准值附近波动范围不大,周期性变化也未出现,表示试验过程符合要求。
4 结语
通过本次分析,得出如下结论:
(1)检出限国标值为0.025,本中心氨氮的测定纳氏试剂分光光度法检出限为0.021,小于国标方法检出限,检出限合格;今后应按检出限0.025使用。
(2)各样品精密度显著性检验合格;从表2可以得出:标准样品(统一标样)测定系统标准差0.006 19、相对误差1.94%,测定结果合格,自制水样系统标准差小于统一标样系统标准差,表明自制水样中不存在影响精密度的干扰因素。
(3)空白值的应用:本次基础实验确定氨氮纳氏试剂分光光度法测定时,空白值在-0.001~0.006 mg/L之间,是合理的,超出测定范围时,应查明原因,重新测定空白样或重新计算方法检出限。
(4)平均加标回收率为103%,符合95%~105%的要求,回收率合格,证明监测数据质量可靠。
氨氮的测定应考虑消除各种干扰因素,使得测定数据更加真实可靠。本次基础试验验证了本中心人员操作、仪器与环境、试剂及纯度均受控,即人员分析水平达到技术要求、仪器操作与维护到位、实验室环境条件符合检测要求、所用试剂纯度与空白值符合国标要求,证明了本中心质量控制到位。
猜你喜欢
化工设计通讯(2022年10期)2022-12-31 20:42:50
波谱学杂志(2022年2期)2022-06-14 09:52:02
云南化工(2021年11期)2022-01-12 06:06:24
World Journal of Clinical Cases(2020年17期)2020-09-18 08:03:24
检验医学与临床(2020年1期)2020-01-10 04:44:22
盐科学与化工(2019年5期)2019-05-21 05:54:04
环境保护与循环经济(2017年12期)2017-07-11 01:46:36
应用海洋学学报(2015年2期)2015-11-22 07:36:40
现代检验医学杂志(2015年1期)2015-02-06 01:59:14
现代检验医学杂志(2015年6期)2015-02-06 01:44:25
