
二硫代水杨酸衍生物对光固化材料性能的影响
2022-10-18 08:19:56鞠小兵李雪纯孙芳
化工学报 2022年9期
鞠小兵,李雪纯,孙芳,2
(1北京化工大学化学学院,北京 100029; 2北京化工大学安庆研究院,安徽 安庆 246000)
引 言
光聚合技术因其高效、环保、节能及室温固化等特点而成为目前发展迅速的绿色技术之一[1-4]。然而,传统光聚合技术所使用的汞灯光源在使用时会产生臭氧和重金属污染,对环境危害极其严重,且使用寿命较短,因此,传统光聚合技术仍具有局限性[5-8]。与汞灯相比,发光二极管(LED)具有寿命长、能耗低、无汞蒸气和臭氧产生、环保安全及出光速度快等优点[9-13],因此,以LED 作为光源的LED 光聚合技术成为目前光聚合技术发展前沿方向[14-16]。众所周知,自由基光聚合由于聚合前单体或预聚物分子之间的范德华作用力距离被聚合后的共价键长度所取代,以及分子间快速交联限制了链段运动,自由体积变小,而不可避免地会产生体积收缩[17-19]。这种收缩会造成材料翘曲变形、内部缺陷等问题,使光固化材料的精细结构和力学性能受到极大影响[20-22]。同样地,LED 光聚合技术也存在与传统光聚合技术相同的体积收缩问题,因此,降低体积收缩也是LED 光聚合技术亟待解决的难题。近年来,本课题组一直致力于降低光聚合体积收缩方面的研究,提出了多种降低体积收缩的手段[23-25]。近期,本课题组将可聚合二硫化合物引入到汞灯作为光源的光聚合体系中,利用光聚合过程中二硫键不断发生“断裂—恢复”的可逆反应来调节体系自由体积,进而达到了有效降低聚合过程中体积收缩的目的[26-28]。然而,前期所设计合成的二硫化合物由于其光吸收波长较短,而无法用于波长较长的LED光固化体系。芳香族二硫化物由于芳香环的共轭效应有利于分子紫外吸收波长红移和降低S—S键键能,因此具有用于LED 光聚合体系的潜力。另外,芳香族二硫化物本身具备一定的光引发能力,具有同时作为LED 光引发剂的可能。因此,希望能设计出在LED 光源发射波长下可进行硫硫键“断裂—恢复”的可逆反应,并能引发丙烯酸酯类单体聚合的可光聚合芳香族二硫化物,使其具有降低体积收缩和引发双重功能,从而改善光固化材料性能,并简化光固化材料配方。
基于上述背景,本文将设计制备一种可聚合二硫代水杨酸类芳香族二硫化物(MAPBS),并研究其光化学性能、降低体积收缩能力以及其对光聚合体系聚合动力学、耐热性能和硬度的影响,以期为二硫化物应用于LED光聚合体系提供理论基础。
1 实验材料和方法
1.1 材料
二硫代水杨酸,分析纯,百灵威化学;1,3-丙二醇,分析纯,萨恩化学;甲基丙烯酰氯,分析纯,天津希恩斯生化科技有限公司;对甲苯磺酸,分析纯,福晨化学试剂有限公司;三乙二醇二甲基丙烯酸酯(TEGDMA),分析纯,上海毕得医药科技有限公司;双酚A 缩水甘油酯(Bis-GMA),分析纯,上海吉至生化科技有限公司;苯基-N-叔丁基硝酮(PBN),分析纯,上海毕得医药科技有限公司;樟脑醌(CQ),分析纯,安徽泽升科技有限公司;甲基丙烯酸二甲氨基乙酯(DMAEMA),分析纯,上海麦克林生化科技有限公司;石油醚、乙酸乙酯、乙腈、二氯甲烷、无水乙醇,分析纯,北京化工厂。
1.2 分析测试仪器
Avance 400 M 型核磁共振波谱仪(NMR),德国Bruker 公司;紫外分光光度计,UV-3600,日本岛津公司;ELEXSYS Ⅱ电子顺磁共振波谱仪,德国Bruker 公司;Nicolet 5700 型实时红外光谱仪,美国Thermo Electron 公司;UVEC-4ⅡLED 点光源照射机,深圳兰普里克公司;HTS-210D 邵氏硬度计,上海精密仪器有限公司;DTG-60AH 热失重分析仪,岛津企业管理(中国)有限公司;LK-10 激光位移传感器,日本KEYENC公司。
1.3 二硫代水杨酸衍生物(MAPBS)的合成
将二硫代水杨酸(3.06 g,10 mmol)、1,3-丙二醇(7.61 g,100 mmol)以 及 对 甲 苯 磺 酸(10.34 g,60 mmol)加入100 ml 三口烧瓶中,搅拌均匀。将反应体系温度升至80℃,保温持续搅拌12 h。实时监测反应,待反应结束后,将反应液冷却至室温,将反应液倒入50 ml 去离子水中,用乙酸乙酯萃取3 次,合并有机层,有机层经无水Na2SO4干燥过夜,而后减压蒸馏除去溶剂得到粗产物。然后,用柱色谱提纯粗产物(洗脱液为石油醚∶乙酸乙酯=2∶1,体积比),得到白色固体状中间产物OH-PBS(3.20 g,产率76%)。取OH-PBS(4.22 g,10 mmol)、三乙胺(2.02 g,20 mmol)置于含50 ml 无水二氯甲烷的100 ml单口瓶中,冰水浴条件下搅拌混合均匀,而后取甲基丙烯酰氯(2.09 g,20 mmol)缓慢滴加到反应体系中,冰水浴保温搅拌4 h,实时点板监测反应,待反应结束后,向反应体系中加入20 ml 饱和碳酸钾溶液猝灭反应。反应液用50 ml去离子水洗涤3次,有机层经无水Na2SO4干燥过夜,而后减压蒸馏除去溶剂得到粗产物。然后,用柱色谱提纯粗产物(洗脱液为石油醚∶乙酸乙酯=4∶1,体积比),最终得到白色固体状目标产物MAPBS(5.02 g,产率90%)。MAPBS合成路线如图1所示。

图1 MAPBS合成路线Fig.1 Synthesis route of MAPBS
1H NMR (400 MHz, Chloroform-d):δ8.09 (dd,J= 7.8, 1.5 Hz, 2H), 7.77 (dd,J= 8.1, 1.1 Hz, 2H),7.49~7.41 (m, 2H), 7.26 (td,J= 7.5, 1.1 Hz, 2H), 6.15(quint,J= 1.2 Hz, 2H), 5.59 (quint,J= 1.6 Hz, 2H),4.53 (t,J= 6.3 Hz, 4H), 4.38 (t,J= 6.2 Hz, 4H), 2.24(quint,J= 6.3 Hz, 4H), 1.97 (t,J= 1.3 Hz, 6H)。13C NMR (100 MHz, Chloroform-d):δ167.35, 166.33,140.42, 136.16, 133.22, 131.52, 127.23, 125.89,125.78,125.55,62.22,61.36,28.11,18.32。
1.4 光吸收性能测试
将MAPBS 配制成浓度为1×10-4mol/L 的无水乙腈标准溶液。使用紫外可见分光光度计测试MAPBS 在200~400 nm 的吸光度(A),而后根据朗伯-比尔定律(A=εcL, 其中L=1 cm,c=1×10-4mol/L)计算出MAPBS在无水乙腈中的摩尔消光系数。
1.5 电子顺磁共振(ESR)谱图测试
将MAPBS 溶解在无水乙腈溶液中配制成1×10-4mol/L 的标准溶液,而后向该标准溶液中加入10 倍的PBN 作为自由基捕捉剂。为了尽量消除环境中的光以及溶剂中少量的氧气对实验的影响,将样品在避光的条件下缓慢通入氮气10 min 消除氧气。在测试的过程中使用0.5 mm 玻璃毛细管吸取适量标准溶液,封端后装入5 mm 石英管中,而后使用波长为365 nm LED 点光源对其照射10 min,最后使用电子共振波谱仪进行测试。
1.6 光聚合动力学测试
以MAPBS 为引发剂,选用单体Bis-GMA/TEGDMA(4∶6 质量比)为活性单体,配制不同浓度的感光液。在洁净干燥的厚度为1 mm 的KBr 盐片上用毛细管均匀地涂抹一层感光液,然后覆盖上一层同样的KBr盐片以隔绝氧气。置于原位红外光谱仪中,而后使用365 nm LED 点光源(光照强度为100 mW/cm2)持续照射样品300 s。光照结束后对双键在1660~1600 cm-1处的红外吸收峰进行积分,通过不同时间下峰面积的不同确定最终的双键转化率,而以时间为横坐标,转化率为纵坐标,即可获得光聚合动力学曲线。通过双键转化率对时间微分即可得到聚合速率曲线。
1.7 聚合物膜的制备
以Bis-GMA/TEGDMA(4∶6,质量比)为活性单体,配制MAPBS 质量分数分别为2.5%、5.0%、7.5%、10.0%的感光液(占单体总质量),感光液置于10.0 mm×0.5 mm×0.2 mm的模具中,在隔绝氧的条件下用波长为365 nm LED光源(光照强度:100 mW/cm2)持续照射10 min固化成膜。同时配制含1.0%(质量)的CQ 和2.0%的DMAEMA 为光引发剂的感光液,在相同固化条件下固化成膜作对比实验。
1.8 聚合物膜体积收缩测试
使用1.6 节配制的感光液,在Keyence LK-G10型激光位移传感器上测试固化后的体积收缩率。首先将待测感光液滴于直径4 mm和高度1 mm的硅胶模具中。使用波长为365 nm 的LED 光源(光照强度:100 mW/cm2)对其照射10 min,激光测微仪记录液膜照射前的高度(l1)和照射后的高度(l2)。通过式(1)得到不同体系的体积收缩率。每组进行三次测试,取平均值。
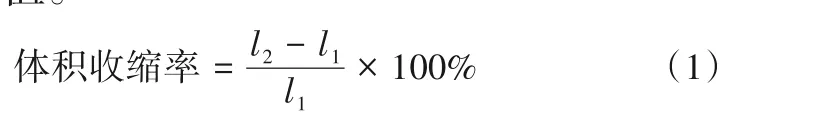
1.9 聚合物膜热稳定性测试
使用热重分析仪(DTG-60AH)测试固化膜的热稳定性,整个测试过程在氮气气氛下进行。测试温度区间为30~800℃,升温速率为10℃/min。
1.10 聚合物膜硬度测试
固化膜的硬度由邵氏硬度计(HTS-210D)测得,样品厚度5 mm,每个样品重复实验五次,取平均值。
2 实验结果与讨论
2.1 MAPBS的紫外可见吸收谱图
MAPBS 的紫外可见吸收谱图及其摩尔消光系数如图2 和表1 所示。从图2 和表1 中可以看出MAPBS 在220、250 和312 nm 附近具有三个强吸收峰,这三个吸收峰是由苯环的共轭结构、丙烯酸酯基中C==C 键和C==O 键以及S—S 键的综合影响所致。在220 nm 处摩尔消光系数最大,达到35700 L/(mol·cm);在250 nm 和312 nm 处的摩尔消光系数也分 别 达 到 了16970 L/(mol·cm)和8380 L/(mol·cm)。值得注意的是,MAPBS 在385 nm 处几乎没有吸收,但在365 nm 处出现了极微弱的吸收,摩尔消光系数为30 L/(mol·cm),说明MAPBS 在365 nm LED照射下有可能发生二硫键的可逆反应和产生自由基。

表1 MAPBS的摩尔消光系数(ε)Table 1 Molar extinction coefficients(ε)of MAPBS

图2 MAPBS在无水乙腈溶液中的紫外-可见吸收光谱(浓度:1×10-4 mol/L)Fig.2 UV-Vis absorption spectra of MAPBS in anhydrous acetonitrile(concentration:1×10-4 mol/L)
2.2 MAPBS的ESR谱图分析
为了探查MAPBS 在365 nm LED 点光源照射下能否发生二硫键断裂产生芳基硫自由基,以PBN 为捕捉剂,对在365 nm LED 点光源照射后的MAPBS的乙腈溶液进行了ESR 测试,结果如图3 所示。从图中可以发现MAPBS 在持续光照条件下产生明显自由基信号,实验测得的曲线与芳基硫自由基拟合曲线相吻合,其超精细裂分常数与文献报道的芳基硫自由基的基本一致(aN=13.8,aH=1.8)[29-30]。这表明在365 nm LED照射下MAPBS可以发生S—S键断裂生成芳基硫自由基。

图3 无水乙腈溶液中MAPBS在365 nm LED点光源照射后的ESR谱图及其拟合谱图(捕捉剂:PBN)Fig.3 ESR spectrum of MAPBS in anhydrous acetonitrile solution after irradiation with 365 nm LED point light source and its fitted spectrum(the capture agent:PBN)
2.3 MAPBS引发单体的光聚合动力学
研究了MAPBS 的引发能力及其浓度对Bis-GMA/TEGDMA 单体光聚合动力学的影响,并与商用光引发剂CQ/DMAEMA 体系做了对比,结果如图4 所示。未外加任何其他光引发剂的情况下,MAPBS 加入量为1.0%(质量分数)时,用365 nm LED点光源持续照射300 s 后体系的双键最终转化率达到55.0%,最大聚合速率为0.69%/s,说明MAPBS 具有引发丙烯酸酯单体聚合的能力。而在同样加入量下,CQ/DMAEMA 体系的双键最终转化率和最大聚合速率都高于MAPBS 体系,分别达到了73.1%和1.12%/s,说明MAPBS 产生的芳基硫自由基活性不如CQ体系形成的氨烷基自由基。当MAPBS在感光液中的含量从1.0%逐渐提高到10.0%时,体系的双键最终转化率由55.0%上升至64.9%,并且最大聚合速率也由0.69%/s提高至1.19%/s。这是由于随着MAPBS 的增加,在持续光照条件下,S—S 键的数量增多,能够产生更多的活性自由基,使得聚合速率明显提高,双键转化率也相应得到提高。
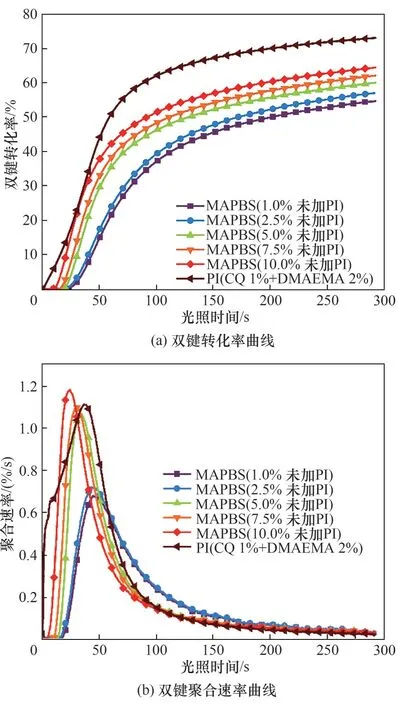
图4 在阻氧条件下MAPBS和CQ/DMAEMA引发单体的光聚合动力学(365 nm LED,光照强度:100 mW/cm2)Fig.4 Photopolymerization kinetics of monomers initiated by MAPBS and CQ/DMAEMA in the absence of oxygen(365 nm LED,light intensity:100 mW/cm2)
2.4 MAPBS对聚合物膜的体积收缩影响
考察了MAPBS 对Bis-GMA/TEGDMA 光聚合体系体积收缩的影响,结果如图5 所示。从图中可以看出,当不添加MAPBS 时,由商业光引发剂CQ/DMAEMA 引发制备的聚合物膜的体积收缩率达到8.10%。在无外加引发剂的条件下,MAPBS 的含量从2.5%增加至7.5%时,聚合物膜的体积收缩率逐步降低,最终可降低至5.07%,这说明MAPBS 的加入可以起到一定的降低体积收缩效果。其降低体积收缩的机制(图6)是MAPBS 分子吸收光能后,较弱的S—S 键立即发生断裂产生芳基硫自由基;然后,一部分芳基硫自由基进攻单体产生初级碳自由基,这个活跃的碳自由基会迅速引发单体进行连锁聚合;而另一部分芳基硫自由基更趋向于重新结合恢复为二硫键。在光聚合过程中恢复的二硫键不断重复着“断裂—恢复”的可逆反应,伴随着这个可逆过程,聚合物网络不断进行着“收缩—膨胀—收缩”的体积调整过程,进而降低了体积收缩[26-28]。然而,当MAPBS 的含量由7.5%提升至10.0%时,聚合物膜的体积收缩率略有升高,这可能是由于当增加MAPBS 的含量时,虽然体系中S—S 键的相对含量得到提升,但也会增加引发活性中心,导致光聚合速率过快,使得S—S键通过“断裂—恢复”可逆过程调节聚合物网络自由体积的效率有所降低。

图5 含有MAPBS聚合物膜的体积收缩率(365 nm LED,光照强度:100 mW/cm2)Fig.5 Volume shrinkage of polymer films with MAPBS(365 nm LED,light intensity:100 mW/cm2)

图6 MAPBS降低体积收缩可能的机理(365 nm LED,光照强度:100 mW/cm2)Fig.6 Possible mechanism of MAPBS reducing volume shrinkage(365 nm LED,light intensity:100 mW/cm2)
2.5 MAPBS对聚合物膜的热稳定性的影响
MAPBS 对Bis-GMA/TEGDMA 光聚合体系热稳定性的影响如图7 和表2 所示。从图7 和表2 中可以看出,随着MAPBS 含量由2.5% 逐渐上升至10.0%,聚合物膜的初始分解温度(T5%)变化不大,但低于不含MAPBS 的MAPBS-0%体系的初始分解温度。这可能是由于由MAPBS 引发制备的光固化膜的双键交联程度较低,固化膜中存在较多的小分子量物质,容易分解、挥发所致。从图7(b)中可以发现含MAPBS的固化膜均会经历两步热失重过程,随着MAPBS 含量的增加第一个最大热失重速率对应温度(Tmax1)由322.4℃降低至310.5℃,第二个最大热失重速率对应温度(Tmax2)由438.0℃降低至429.4℃,表明随着MAPBS 含量的增加聚合物膜的热稳定性降低。这主要是由于S—S 键以及C—S 键的键能相比于C—C 键和C—O 键的键能较低,当温度上升到一定值后S—S键会率先发生断裂,导致聚合物中交联网络被破坏,并且产生较多的小分子片段,因此,体系随着MAPBS 含量提高,固化膜中S—S 键随之增加,固化膜的热稳定性略有降低。

图7 MAPBS体系固化膜的热失重及失重速率曲线Fig.7 Thermogravimetric curves and mass loss rate curves of polymer films with MAPBS
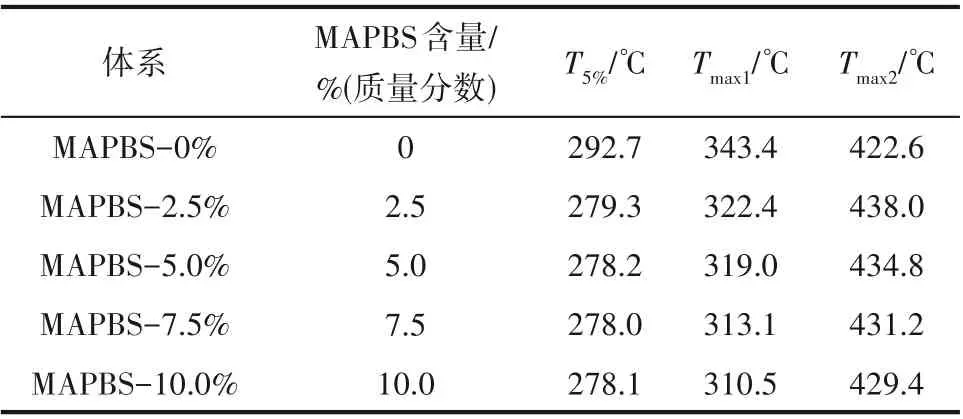
表2 MAPBS体系固化膜的热失重数据Table 2 Thermogravimetric data of polymer films with MAPBS
2.6 MAPBS对聚合物膜的硬度的影响
MAPBS 对Bis-GMA/TEGDMA 光 聚 合 膜 硬 度 的影响如图8 所示。由图中可知,含有MAPBS 的各个体系固化膜的硬度都小于不含MAPBS 的MAPBS-0%体系,这是由于含有MAPBS 的固化程度相对较低且含有较多柔性的S—S 键和C—S 键所致。另外,含有MAPBS 的各个体系固化膜,当MAPBS 的含量从2.5%上升至10.0%时固化膜的邵氏硬度由62.23 HD 上升至71.90 HD,固化膜硬度整体呈现上升状态,这可能是由于随着MAPBS 含量增加,体系聚合程度和刚性的苯环含量都随之增加,使聚合物膜的硬度有所提高。
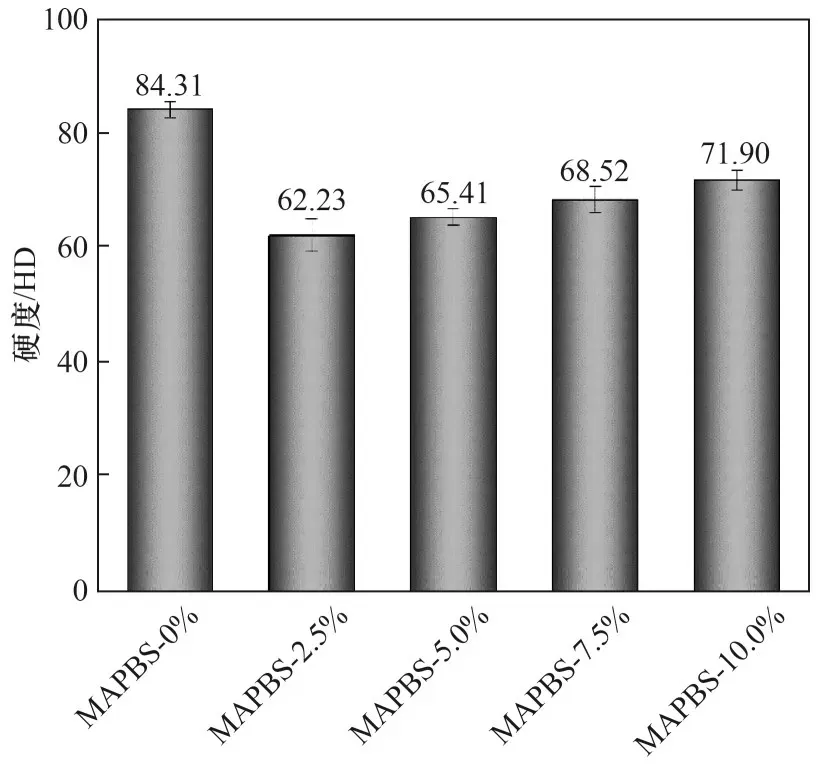
图8 含有MAPBS体系聚合物膜的硬度Fig.8 Hardness of polymer films with MAPBS
3 结 论
(1)在365 nm LED 光源照射下,MAPBS 由于可以发生S—S 键的断裂产生具有引发能力的芳基硫自由基而引发甲基丙烯酸酯类单体聚合。当MAPBS在感光液中的含量从2.5%增至10.0%时,光聚合体系的双键转化率和聚合速率随之增加,分别达到64.9%和1.19%/s。
(2)MAPBS 具有降低丙烯酸酯类光聚合体系的体积收缩的能力。与空白体系体积收缩(8.10%)相比,含有MAPBS 的聚合体系的体积收缩可以降至5.07%。随着MAPBS 含量从2.5%增至10.0%时,聚合物膜的体积收缩呈现出先增加后降低的趋势。MAPBS兼具引发和降低体积收缩的双重功能。
(3)随着MAPBS 含量的增加,聚合物链上键能低的S—S 和C—S 键以及刚性的苯环也随之增多,综合影响使聚合物膜的热稳定性略微降低,但硬度稍微有所提高。
猜你喜欢
好日子(2022年6期)2022-08-17 07:17:06
机械工业标准化与质量(2022年6期)2022-08-12 02:07:48
影像技术(2019年5期)2019-09-10 07:22:44
传感器世界(2019年11期)2019-02-17 01:17:35
中国调味品(2017年2期)2017-03-20 16:18:13
合成化学(2015年2期)2016-01-17 09:04:21
化工进展(2015年6期)2015-11-13 00:27:23
中国塑料(2015年10期)2015-10-14 01:13:13
中学化学(2015年2期)2015-06-05 07:18:13
化工生产与技术(2014年3期)2014-02-27 13:41:42
