
氨电氧化催化剂及其低温直接氨碱性膜燃料电池性能的研究进展
2022-10-18 08:16方辉煌程金星罗宇陈崇启周晨江莉龙
化工学报 2022年9期
方辉煌,程金星,罗宇,陈崇启,周晨,江莉龙
(福州大学石油化工学院,化肥催化剂国家工程研究中心,福建 福州 350002)
引 言
氢是公认的绿色高效的清洁能源,然而,氢气在常温常压下能量密度低,仅为0.0108 MJ·L-1,需加压至70 MPa 方可将其提升至6 MJ·L-1,从而进行高压存储。同时,氢气易燃易爆,在长距离运输和供应网络等方面存在较多的技术挑战[1-3]。如何实现氢气的安全存储和高效利用是解决氢能大规模利用的关键。
利用化学手段将氢气进行液态存储是解决这一关键难题的有效途径[4-6]。相比于传统的甲醇、乙醇、甲酸、碳氢化合物等含氢化合物,氨是一种无碳的富氢能源载体,具有能量密度高(13 MJ·L-1),含氢量高(17.7%)等特点,常温下易液化,运输和储存基础设施成熟,且终端无碳排放,有助于实现碳达峰和碳中和[7-8]。单位体积下,液氨的含氢量和能量密度分别为液态氢的1.7 倍和1.5 倍以上。以氨作为富氢燃料用于能量供给系统可直接实现氢气的高效存储和能源的区域再分布,是理想的能源路线之一。在氨能利用中,主要有氨内燃机、间接氨燃料电池(氨制氢-氢燃料电池)和直接氨燃料电池等[9-10]。在直接氨燃料电池技术中,氨碱性膜燃料电池(NH3alkaline exchange membrane fuel cell, NH3-AEMFC)可在低温下(<100℃)工作,相比于高温固体氧化物燃料电池具有体积小、启动快等优势,不仅可实现其在瓦级至百兆瓦级的发电/动力应用场景下的灵活供电,还具有优异的“氨-电”理论转化效率;因此,NH3-AEMFCs是低温下实现氨能高效利用最可行的途径之一[11]。图1 示出了直接氨碱性膜燃料电池的组成及工作原理。其中,氨在阳极发生氨氧化反应(ammonia oxidation reaction, AOR),氧在阴极发生还原反应(oxygen reduction reaction,ORR),阴极产生的OH-通过碱性离聚物膜,参与阳极AOR 生成水和氮气,从而将氨的化学能转化为电能。具体反应方程式如下:
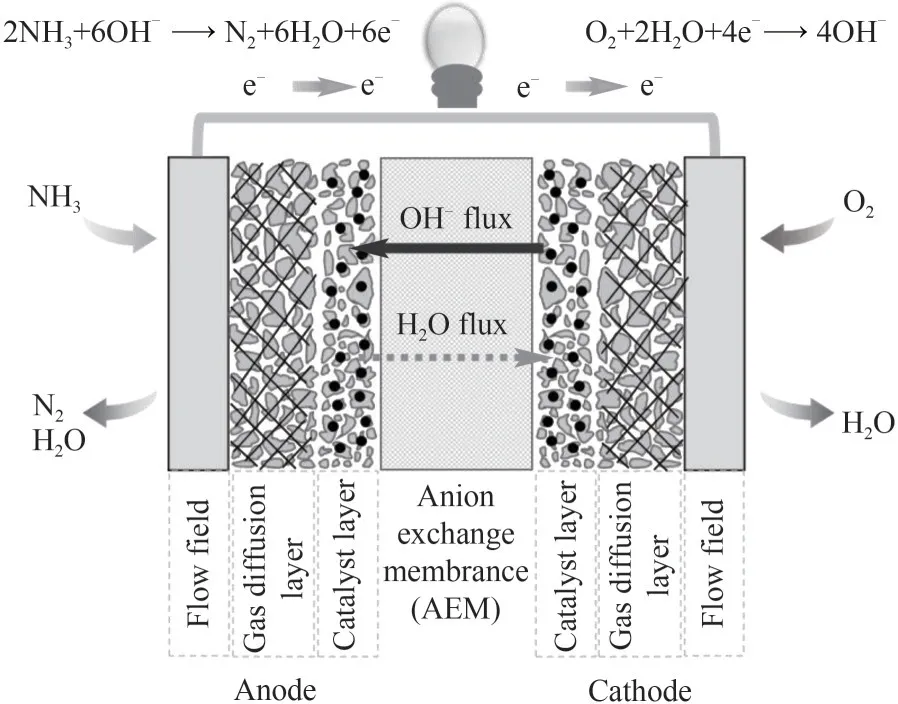
图1 直接氨碱性燃料电池示意图Fig.1 The schematic diagram of NH3-AEMFC
阳极半反应
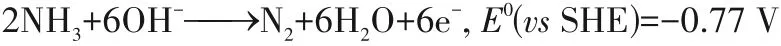
阴极半反应
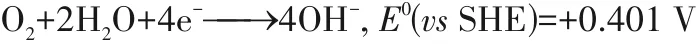
相比于氢氧燃料电池的阳极氢氧化反应,氨氧化反应动力学缓慢,需要较高的过电位(>0.4 V);同时该过程生成的中间物质(*N、*NO)强烈吸附在催化剂表面占据活性位点,从而导致催化剂中毒现象的发生[8,12-14]。因此,如何设计高效、廉价、稳定的催化剂是实现清洁氨能利用的重点和难点。本文将讨论氨氧化的反应机理,重点综述氨电化学氧化中的催化剂研究进展,总结直接氨碱性燃料电池的性能,并对未来的研究方向进行总结和展望,旨在为深化氨氧化反应的构效关联研究和催化剂设计提供思路。作为直接氨碱性膜燃料电池中的阴极氧还原反应,与氢氧燃料电池体系一致,限于篇幅本文不作赘述。
1 氨电化学氧化反应机理
在直接氨碱性燃料电池中,阳极发生AOR,该过程反应动力学缓慢且存在催化剂中毒现象。因此,AOR 机理的研究有助于合理设计高效稳定的电催化剂,为进一步提升NH3-AEMFC 的性能及其商业应用提供了可能。目前,碱性电解质中阳极Pt 基催化剂的AOR 反应机理主要分为两种途径,如图2所示。第一种途径是Oswin 等[15]在1963 年提出的逐步脱氢机理(简称N+N 机理)。该反应机理主要过程如下:氨首先吸附在Pt 催化剂活性位点上,然后与吸附活化的OH-反应并逐步失去氢原子,最后吸附的两个氮原子(*N)相互二聚形成N2分子。基于Tafel斜率(39 mV·s-1)和Langmuir 等温线模型的分析,该理论认为在低电流情况下,*NH2脱氢至*NH 过程为反应的决速步骤;而在高电流条件下,*N 二聚形成N2分子为反应的决速步骤。*N 物种在过渡金属表面的结合能达394 kJ·mol-1以上,从而导致难以结合形成N2
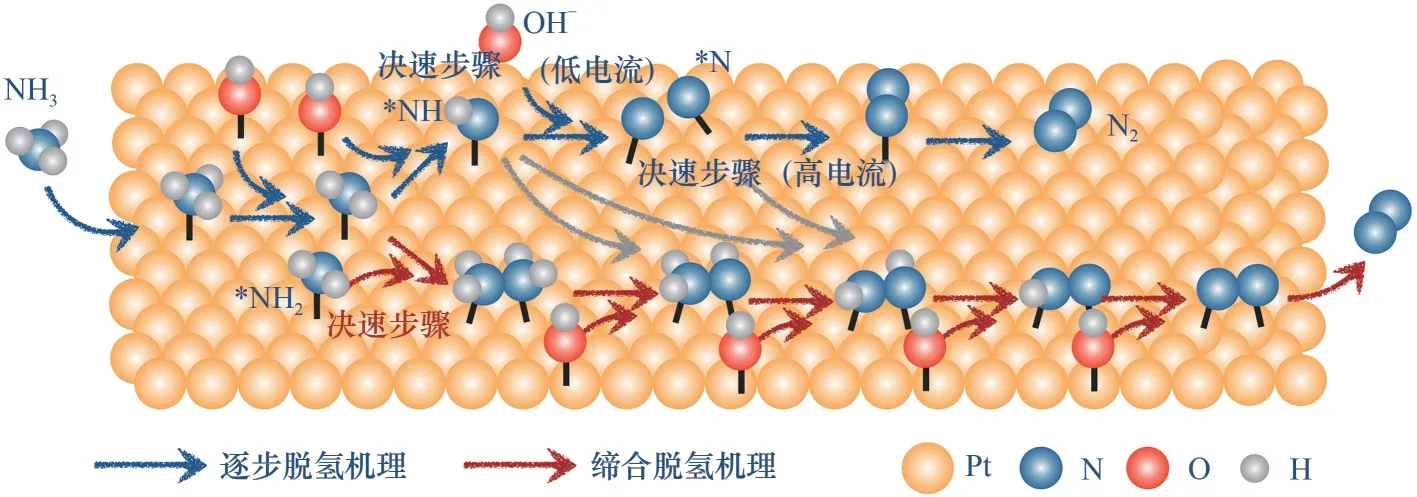
图2 氨氧化反应机理Fig.2 Reaction mechanism of AOR
[16-17]。第二种途径是Gericher 等[18]在1970 年提出的缔合脱氢机理(简称G-M 机理)。该机理为:*NH3逐步脱氢生成不同的吸附中间体(*NH、*NH2),然后中间体之间相互重组形成*N2Hy(y=2~4)物种,最后脱氢生成最终产物N2。在此机理中,*NHx的二聚反应是决速步骤,吸附的*OH-可能在AOR机理中起重要作用。相比于G-M 机理的二聚过程,完全脱氢的*N 物种在催化剂表面的吸附能高,容易毒化电极催化剂表面,使催化剂失活。
与N+N机理相比,G-M机理被众多研究者通过原位表征实验[19-23]和理论计算[24-26]验证。de Vooys等[19,27]采用微分电化学质谱(differential electrochemical mass spectrometry, DEMS)和循环伏安(cyclic voltammetry,CV)方法研究了AOR 的反应中间物种,发现*NH和*NH2物质是活泼的中间物种,并进一步证实*N 物种在催化剂表面的毒化作用。Vidal-Iglesias 等[20]通过表面增加拉曼光谱(surface-enhanced Raman spectroscopy, SERS)检测到叠氮化物阴离子(N-3),这主要源于*N2H4与氨的反应,进而中间物种N-3进一步参与AOR反应。Matsui等[22]采用原位衰减全反射红外光谱(attenuated total reflection infrared spectroscopy,ATR-IR)研究了Pt 电极在碱性水溶液中的AOR 过程,观察到吸附态的*NH3、*N2H4和桥接*NO 的三个特征吸收峰,其强度随电位的变化而变化。这是首次在Pt电极上检测到*N2H4的存在,进一步佐证了G-M机理。Ye 等[23]用原位红外光谱(Fourier transform infrared spectroscopy, FTIR)监测AOR 的反应过程,在低电位下可以清晰地观察到*NH2和*N2H4物种的特征红外峰,与G-M 机理相符合。此外,在较高的电位下,N2O、NO2等其他中间物质被检测到,而这些物质的形成会影响N2的选择性。表1 列出了AOR反应中研究者检测到的反应中间物种及采用的原位表征技术手段。这些技术的使用有助于研究AOR 的反应机理,深入催化剂的构效关系探究,为设计高效稳定的AOR电催化剂提供理论指导。

表1 原位表征技术观察AOR过程产生的中间物种Table 1 The intermediates of AOR process by in situ characterization techniques
另一方面,密度泛函理论(density functional theory, DFT)计算是研究AOR 反应途径和机理的重要手段。Skachkov 等[24]在实验探究的基础上,通过对Pt低指数晶面上AOR进行仿真模拟发现:在低电位区下(<0.50 VvsRHE),*NH2、*N2Hx+y和*N 是中间物质,AOR 遵循G-M 机理;在较高电位下(>0.50 VvsRHE)遵循N+N 机理。但是,关于AOR 遵循N+N 还是G-M 机理仍存在争论。Diaz 等[26]的DFT 计算表明N2的形成受氨吸附中间体(*NH2)的耦合作用控制。Katsounaros 等[30]也报道了相似的结果。他们通过实验和理论计算表明N2是通过*NH+*NH 耦合步骤形成的,这与G-M 机理相符合。然而,Li 等[31]认为*N 表现出更稳定的热力学特性,更易形成N—N键。这两种机理可能在反应热力学和动力学的指导下存在竞争关系或共存关系[18,32]。除了金属Pt 催化AOR 体系,Herron 等[25]也利用DFT 计算研究了一系列过渡金属对AOR的催化活性,同时考虑了G-M和N+N 机理途径。基于活化能和热力学的计算,他们建立了AOR催化活性与金属催化剂对N的结合能关系图。图3 示出了Rh 和Ir 对N 结合能较高会使N—N 结合变得更加困难;而Cu 在理论上表现出对AOR的高活性,但它与N原子的结合太弱,实验也进一步证明这一理论结果[25,33]。此外,根据Sabatier 原则,最佳AOR的催化剂应该在Pt和Cu之间有个中等N的结合强度[34]。因此,催化剂的合金化有望为开发和设计高效AOR催化剂提供新的思路。
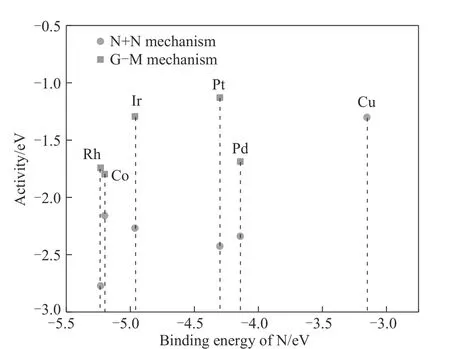
图3 AOR催化活性与金属催化剂对N的结合能关系[25]Fig.3 The relationship between the AOR activities and the binding energy of metal catalyst to N[25]
此外,一些研究者提出了新的补充和见解。Novell-Leruth 等[35-36]利用平面波周期DFT 计算研究Pt 晶面上吸附氨及脱氢中间物质的几何形状和位置偏好关系,发现Pt(100)上的*NH3和*NH2比在Pt(111)上更稳定,这表明AOR 是结构敏感反应;在Pt上最稳定的吸附构型为*NH3端位,*NH2桥式,*NH、*N 的中空位。Daramola 等[37]计算了AOR 过程中可能的反应中间物质在Pt(111)晶面吸附能,并预测了它们之间关系:N >NH >NH2>OH>N2H3>N2H >N2H4>NH3>H2O >N2。在此基础上,他们报道双金属Pt-Ir 催化剂,发现AOR 在Pt 上遵循G-M 机理,在Ir 上遵循N+N 机理;PtIr 二元合金中Ir 的活性位点吸引*NH、*N,这有助于*N2H4在Pt的活性位点上反应形成N2[32,38]。Katsounaros 等[39]研究溶液pH 对氨在Pt(100)上的电催化氧化的影响,并提出解耦的质子-电子转移过程。此外,Peng 等[40]认为含氧物种(*O、*OH)与氨的中间物质存在竞争关系,进而抑制AOR反应;同时,他们对非水介质中的AOR进行了DEMS研究,发现没有含氧物种的存在,Pt表面对AOR保持良好活性。最近一项研究中,Yang 等[41]基于DFT 分子动力学模拟与自由能相结合方法,研究了Pt(100)表面上AOR 过程中*NH3向N 的逐步脱氢过程,他们发现吸附*OH 辅助脱氢反应对电势几乎不敏感,而溶液中存在的大量OH-对电势有依赖性,电势越低,这种反应的势垒越高。这表明氨脱氢反应中*OH 是反应物质,而不是在水中的OH-。综上,原位表征技术和DFT计算揭示了AOR 过程的中间物质、中毒物质以及金属对N 的结合能,对于如何合理设计催化剂具有重要指导意义。
2 氨电化学氧化催化剂
目前,常用的AOR 催化剂有以Pt基催化剂为代表的贵金属催化剂和以Ni、Cu等为代表的非贵金属催化剂。这些催化剂的结构特性直接影响AOR 的催化性能。目前,催化剂活性差,贵金属催化剂成本昂贵,催化剂稳定性差等是限制NH3-AEMFC 阳极催化剂商业化应用的主要因素。如何提高催化剂性能,并大幅降低成本,已成为众多科研工作者的关注热点。为此,本节简述近些年来在阳极AOR催化剂领域的研究进展。
2.1 贵金属催化剂
Pt 基催化剂是最为常见的用于AOR 的贵金属催化剂。然而,储量低、价格昂贵等限制了其大规模应用。如何提高Pt 的原子利用率及单位活性是研究Pt 基催化剂的重点。如图4 所示,当前Pt 基催化剂的研究主要分为以下几类:(1)对Pt纳米颗粒进行结构和晶面调控,使其暴露更多高活性位点,提高Pt 基催化剂的单位活性;(2)利用载体调控,提高Pt 纳米颗粒的分散性,并利用载体与金属间的相互作用,一方面提高催化剂活性,另一方面降低Pt 的负载量;(3)引入其他金属和助剂等,构筑双金属或多金属合金催化剂或核壳结构催化剂等,调控Pt 基催化剂的电子结构,并利用多活性位点的协同作用,提高Pt基催化剂性能。
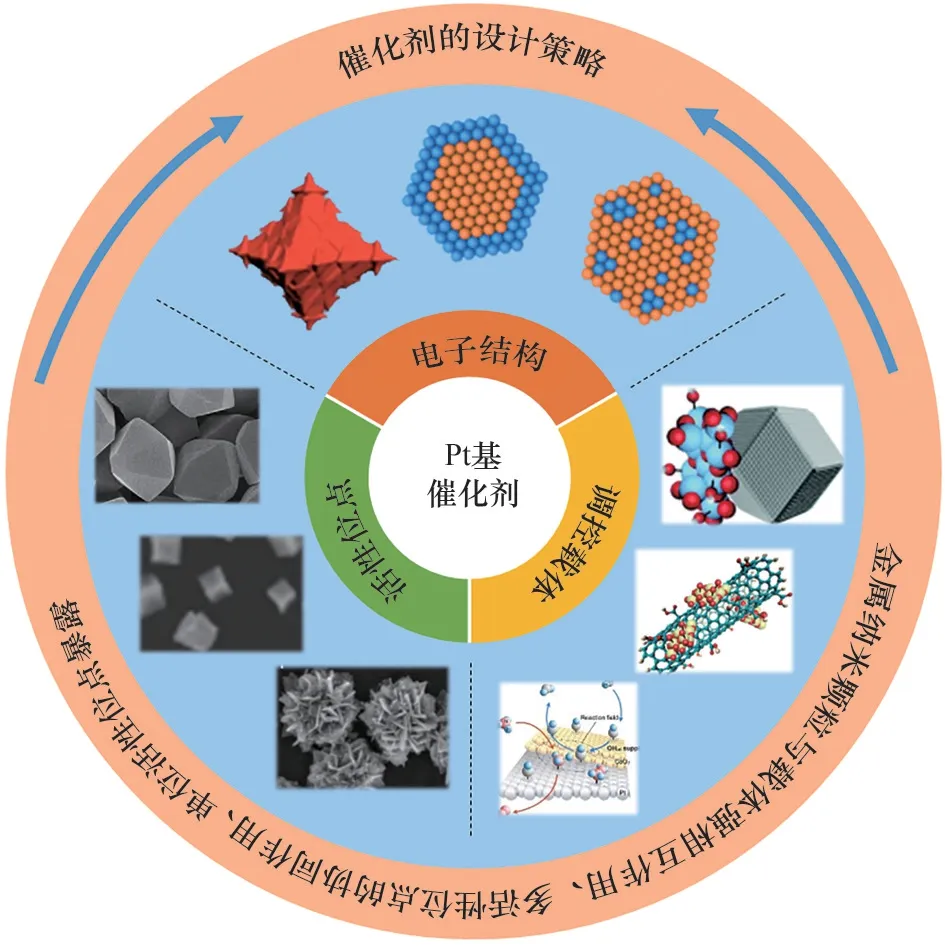
图4 提高AOR活性的途径Fig.4 Scheme of general approaches to improve activity for AOR
氨在Pt 表面氧化是一种结构敏感反应过程,其AOR 的电催化性能高度依赖Pt 的纳米颗粒形貌和暴露晶面。AOR 反应更倾向于在Pt(100)晶面上发生,而其他晶面[(100)、(111)]催化活性较低[35,42-45]。因此,合成择优取向(100)晶面或具有一定形貌的铂纳米颗粒是提高AOR 活性的可行方法。Zhang 等[46]采用浸渍还原法制备碳载立方Pt 纳米晶体(PtNC/C),并与商业Pt/C 对比,其AOR 峰值电流密度提高5 倍,并表现出更好的稳定性。Fu 等[47]通过水热还原法合成了暴露(100)晶面Pt纳米立方体,因Pt纳米颗粒的择优取向和尺寸效应,其电催化活性和持久性显著优于商业Pt黑催化剂。类似地,Zhong等[48]报道了(100)晶面取向的PtIr 纳米立方体,表现出优异的AOR 活性(1.32 mA·cm-2),远高于PtIr 纳米颗粒(0.2 mA·cm-2),也高于纯Pt 立方体(1.05 mA·cm-2)。这表明Ir 的掺入不但没有破坏Pt(100)的活性位点,而且两者之间表现出协同催化效应。此外,Hu[49-51]通过控制电沉积的电流密度和电位制备出不同形状(片层状和花状)的Pt纳米颗粒。相比于球形Pt纳米颗粒,具有花状或片层状结构的Pt 纳米催化剂具有更高的电活性表面积,从而表现出更高的AOR活性。
另一方面,利用载体担载活性金属纳米颗粒可以显著提高催化剂的性能。载体影响着活性金属的分散度、粒径以及电导率等,同时还能与活性金属形成金属-载体相互作用,进而影响催化剂活性、燃料电池的性能及耐久性。在电催化反应中,理想的载体应满足如下要求:(1)良好的电导率;(2)活性中心与载体具有良好的相互作用;(3)大比表面积;(4)在氨电氧化条件下的高电化学稳定性[52]。目前常见的用于AOR 反应的载体有:碳基材料[53-57]、氧化物[58-59]以及复合载体[13,60-61]。Ribeiro 等[53]制备了氮掺杂碳(N-Cx)载体担载Pt 纳米颗粒,并对其进行AOR活性评价。实验结果表明Pt/NCx的AOR 活性高于Pt/C,其中Pt/NC5的峰值电流密度比Pt/C高161%。这表明载体上的氮的掺杂促进了AOR过程。Ntais等[58]合成了Pt/NiO、Pt/MnO2催化剂,实验结果显示,Pt/NiO 表现出最低的起始电位和最高的电流密度,且峰值电流密度向低的过电位偏移,这主要归因于Pt与NiO 之间的电子金属载体相互作用,以及NiO 载体向反应位点提供额外的*OH。最近研究报道,氧化物与碳材料组合的复合载体用于AOR 反应研究并表现出良好的催化性能。Yan 等[13,60]详细探讨了单一载体和复合型载体对AOR 反应影响。首先把Pt 负载在单一载体和复合的载体上,发现二氧化硅(SiO2)与功能化的碳纳米管(CNT-COOH)之间的协同作用显著提高了AOR 活性;紧接着改变金属氧化物(ZrO2、CeO2、TiO2),也同样证明SiO2-CNT-COOH 活性最好。这是因为多孔SiO2具有较大的比面积(>400 m2·g-1)且易于吸附金属离子,此外还向活性位点提供丰富的*OH;而CNT-COOH 可以提高导电性能,增强结构完整性。然而负载在SiO2-CNT-COOH上的纳米粒子存在分散不均匀情况。基于此,由于CeO2中的Ce3+可以作为路易斯碱修饰反应物和中间物质,他们研究了CeO2和不同碳载体的复合,发现CeO2与咪唑啉分子筛骨架-8(ZIF-8)复合的载体有助于金属纳米粒子的分散。此外,助剂金属氧化修饰Pt 基催化剂也可以显著提高AOR 活性。Eguchi 等[28,62-64]在该方面做了系统的研究工作。他们用稀土氧化物(CeO2、Y2O3、La2O3和Sm2O3)修饰Pt催化剂,发现稀土氧化物添加改善了*OH 对Pt 位点供应能力;其中,CeO2修饰的Pt 催化剂活性最好,峰值电流密度是Pt 催化剂的3.5 倍。为进一步论证,他们采用ATR-FTIR 研究了CeO2修饰Pt 和离聚物涂覆Pt 催化剂上的电极表面物种,以阐明电化学反应过程中*OH 吸附行为的变化。在空白KOH 溶液中ATR-FTIR 测量结果表明,CeO2添加剂影响Pt对*OH 的吸附行为,但不会改变Pt-Had键的强度或吸附焓;在AOR中也观察到*OH吸附行为的变化。
Pt 基催化剂的合金化可以显著提高AOR 电催化性能。通过Pt合金化,可以对AOR性能进行精细调控,加入第二金属组分一方面改变了催化剂表面的电子结构性质,另一方面与Pt 产生协同作用,从而提高AOR 的活性和稳定性。Baranova 等[65]研究了碳负载PtM(Pd,Ir,Sn 等)双金属纳米粒子在AOR 反应中的性能。结果表明,Pd 的引入降低起始电位,但稳定性变差;Pt(SnOx)3电流密度与Pt相似,但催化剂的稳定性得到了改善;而Pt7Ir3纳米粒子具有较高的催化活性和稳定性。这主要是由于在Pt 表面引入Ir,使Pt 表面的电子结构发生变化,从而改变PtIr对反应物和中间物质的吸附能力,从而提高了PtIr的催化活性[66]。Imbeault 等[67]制备一系列PtxIr100-x薄膜用于AOR 研究,结果发现与纯的多晶铂相比,这些合金在AOR 反应中具有更好的抗毒性,其中Pt89Ir11合金表现出的性能最优。PtIr 催化剂表现出的高活性和稳定性与其表面的电子效应和协同效应有关。Morita 等[68]成功合成了PtIr 核壳纳米颗粒(Pt-Ir/MWCNT),并进行动力学研究分析。实验结果表明Pt-Ir/MWCNT 氨氧化表观速率高于Pt/MWCNT,表明Pt 电子受到Ir 核原子影响。Chan 等[69]采用湿化学法合成了(100)面取向Pt-M 纳米立方体(M = Fe, Co, Ni, Zn)和PtIr 纳米立方体,与商业的Pt/C 催化剂相比,过渡金属元素的加入均提高AOR 活性,其中PtIr 电催化活性最好,PtZn 其次。活性的提高归因于过渡金属原子优先吸附羟基,有助于暴露更多的Pt 位点。因为羟基会与氨竞争Pt活性位点,抑制AOR[40,69]。此外,在Pt 基二元催化剂基础上加入另一种金属,这类三元体系的电催化剂对氨具有很高的催化活性和选择性,不仅提高AOR的峰值电流密度,而且起始电位也得到进一步下降[70]。目前研究过的三元催化剂有PtIrRh[71]、PtIrZn[72-73]、PtPdRh[74]、PtIrNi[13]、PtIrCu[75]以 及PtCuRu[76]。Wu 等[73]通过超声-辅助还原的方法制备出PtIrZn纳米颗粒,且均匀分散在由CeO2和ZIF-8组成的二元复合载体上(PtIrZn/CeO2-ZIF-8)。与商业的PtIr/C相比,最优比例的PtIrZn具有较高的峰值电流[31.8vs25.1 A·(g PtIr)-1]、较低的起始电位(0.35vs0.43 V)以及较低的活化能(约36.7vs50.8 kJ·mol-1)。DFT 计算结果表明:Zn 原子加入PtIr 合金中可以调节PtIr 体系(100)晶面的电子结构,促进*NH3脱氢过程。Lin 等[75]采用溶剂热法成功合成了具有{553}、{331}以及{221}高指数面的八面体PtIrCu 纳米枝晶。该纳米结构展现出卓越的催化性能,峰值电流密度达到122.9 A·(g PtIr)-1,经过2000次循环加速稳定性测试后,峰值电流只下降了17.7%。
2.2 非贵金属催化剂
Pt 基贵金属催化剂资源稀缺、价格高昂等缺陷,使其商业实用性受到限制。因此,开发非贵金属催化剂至关重要。Ni 基催化剂是应用于AOR 反应中的潜在催化材料。通过DFT计算,Ni的AOR起始电位为0.33 V,与Pt(0.28 V)非常接近,但AOR 动力学受到联氨(*N2H4)形成的高能垒限制(1.39 eV)。早期的研究结果表明纯Ni 对AOR 的活性较低[77-78]。然而在碱性溶液中进行电位循环可以激活该物种并形成Ni(OH)2保护层,之后通过氧化反应将其转化为NiOOH[79-80]。目前,关于Ni 电极上AOR 机理存在两条路径:(1)间接电子转移机理[81];(2)直接电子转移机理[82]。
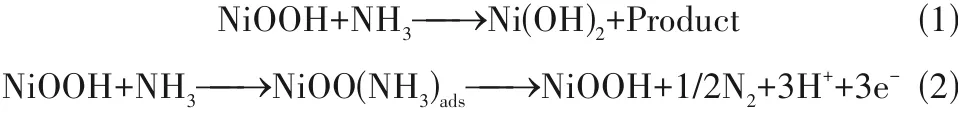
间接电子转移[反应式(1)]和直接电子转移[反应式(2)]途径的反应速率取决于电解液中氨的浓度。在低氨浓度下,主要以反应式(1)发生,而在高浓度下则以反应式(2)为主导。当所有活性表面位点都达饱和时,氨氧化反应将遵循反应式(1)的过程[83]。然而,Ni材料的AOR 催化剂存在腐蚀问题和含氧氮物种产生,导致整体效率偏低。为了克服这些问题,许多研究者通过Ni纳米颗粒表面结构调控以及引入其他金属形成合金并调控形貌来提高镍基催化剂耐腐蚀和N2选择性[84-85]。例如Tao 等[85]通过水热反应和电化学活化,在碳纤维布电极上直接合成具有分层的混合NiCu 层状氢氧化物纳米线(图5)。改变Ni/Cu 比例并获得最佳比例为0.8∶0.2。然后,电化学活化处理产生NiCu 混合氧化产物作为活性位点。对纯Ni(OH)2和Ni0.8Cu0.2进行CV测试,观察有相同的起始电位,但Ni0.8Cu0.2LHs 的电流密度急剧增加,在0.55 V 时 达 到35 mA·cm-2,是 纯Ni(OH)2(5 mA·cm-2)的7 倍。此外,为了做比较,其他第一行过渡金属Cr、Mn、Fe、Co、Zn 也分别作为掺杂剂制备了Ni 基LHs,但在这些元素中,Cu 是唯一能显著提高AOR 催化活性的元素。接着,他们又在碳纸上电沉积制备NiCu 双金属催化剂用于AOR 和氨电解过程。NiCu双金属比Ni或Cu表现出更高的催化活性;以NiCu负载于碳纸作为阳极进行氨电解,其除氨效率达到了80%,库仑效率达到了92%[85]。NiCu/C作为阳极催化剂用于NH3-AEMFC上是可行的[86-87]。

图5 NiCu在碳纤维布上生成和电化学激活流程Fig.5 Catalyst preparation of NiCu LH nanowires and electrochemical activation of the electrode
He等[88]报告NiCo2N纳米片可以实现非水溶液中的AOR的高电催化活性。CV 曲线表明NiCo2N纳米片的AOR起始电位为0.55 V,比Pt/C催化剂低0.25 V。紫外可见光谱和DFT计算显示,在NiCo2N纳米片中,d带收缩使得氮结合强度适中,更有利于AOR中的决速反应步骤,包括NH+x阳离子自由基的生成,以及对氨的吸附和最终*N2的解吸,从而得到更优的催化性能。Zott 等[89]报道了[(bpyPy2Me)Fe(MeCN)2]2+分子在AOR 中具有优异的性能,其转换次数(turnover number,TON)可高达149。他们发现通过配体调控,实现了对Fe金属原子价态调控,同时调控配体的结构,改善催化剂稳定性和催化反应活性。表2 总结了近些年来研究者们报道的AOR 催化剂,以及它们的本征活性(起始电位)和反应速率(峰值电流);虽然随着研究的深入,AOR 催化剂性能已经有长足的进步,但由于实验条件如电解液溶液和扫描速率等的差异,其相互之间的性能较难比较,更需要将其应用于单电池性能测试中。

表2 AOR在贵/非金属催化剂上的性能Table 2 Activity of AOR over non-/noble metallic catalysts
3 低温直接氨碱性膜燃料电池性能研究进展
在低温直接氨碱性膜燃料电池中,功率密度如峰值功率密度和某电位/电流下的功率密度是衡量与评价单电池性能的重要指标。碱性溶液AOR 测试研究有助于高效筛选出用于NH3-AEMFC 的阳极催化剂,但难以考察传质、导电等对低温直接氨碱性膜燃料电池性能的影响。因此,研究者需要进一步将催化剂用于组装膜电极(membrane electrode assembly,MEA),并进行单电池性能测试。如图6 所示,单电池结构主要由端板、集流板、流场板、密封片及MEA组件构成。随着碱性阴离子交换膜工艺的不断提升和改进,在相对较高的温度下显示出优越的阴离子电导率,这为NH3-AEMFC的规模化应用提供了可能[96]。Yan等[11,97]以耐氨的Acta 4020非贵金属催化剂作为阴极催化剂,商业的PtIr/C作为阳极以及聚芳基哌啶的氢氧交换膜[poly(arylpiperidinium),PAP],制备了面积为5 cm2的膜电极组件进行测试,在温度为80℃,3 mol·L-1KOH 条件下,峰值功率密度为135 mW·cm-2。接着,他们进一步研究了背压和阴极催化剂(Acta 4020、Pd/C 和Pt/C)对NH3-AEMFC 性能和耐久性的影响[98]。以Acta 4020 为阴极的DAFCs 可以达到390 mW·cm-2的峰值功率密度,但在300 mA·cm-2下只运行11 h。而阴极为Pd/C 时具有中等的性能,峰值功率密度为304 mW·cm-2,但耐久性大幅度提高,在300 mA·cm-2下可连续工作36 h,缓慢衰减率为1 mV·h-1。类似地,Wu等[73]也采用了PAP 膜,以PtIrZn/SiO2-CNT-COOH 作为阳极,Acta 4020 作为阴极,其单电池的峰值功率密度为314 mW·cm-2,远 高 于 PtIr/SiO2-CNT-COOH(282 mW·cm-2)和PtIr/C(241 mW·cm-2)。Achrai 等[99]报道了阳极燃料溶液不含KOH 的直接氨燃料电池。他们以Pt1Ir10/C 作为阳极材料、Ag 作为阴极及Tokuyama膜制备成MEA用于有KOH和无KOH存在的溶液测试。研究发现在120℃,16 mol·L-1NH3和无KOH 的条件下峰值功率密度达到180 mW·cm-2。此外,Tao 等[100]在阴极与阳极均选择了非贵金属材料(铬修饰镍作为阳极,二氧化锰作为阴极)以及阴离子交换膜[chloroacetyl poly poly(vinyl alcohol),CPPO-PVA]制备成MEA,室温下峰值功率密度为11 mW·cm-2。表3 列出了已报道的NH3-AEMFC 性能(峰值功率密度)结果。综上,NH3-AEMFC 性能明显得到提升,但其峰值功率密度和最大电流密度仍低于低温碱性膜氢燃料电池,表明阳极催化缺乏高活性和稳定性,而对MEA 工艺、反应器流道及水热管理亦需进一步开展针对性优化研究工作。
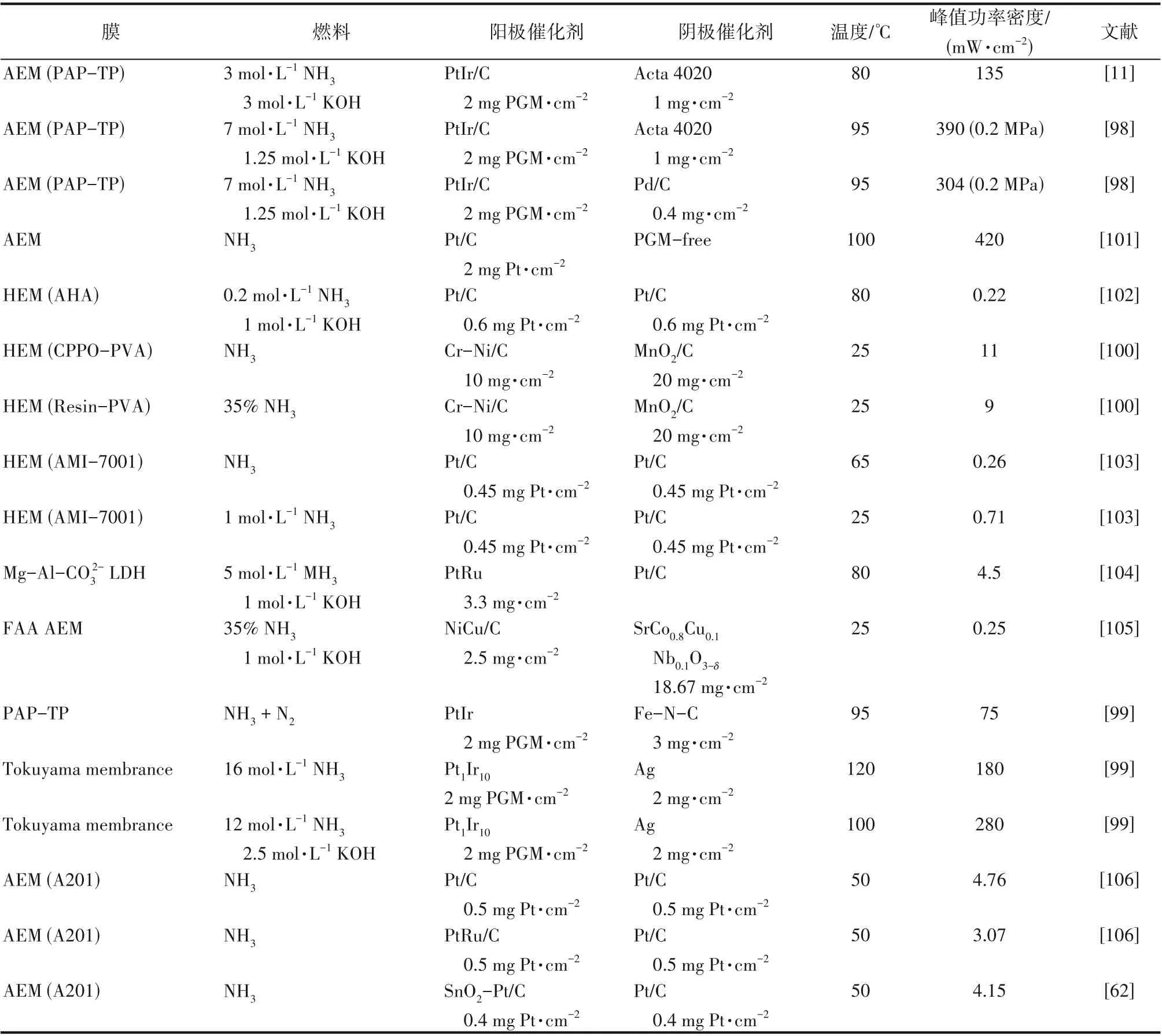
表3 碱性膜直接氨燃料电池性能Table 3 AEM-DAFC performance with various catalysts
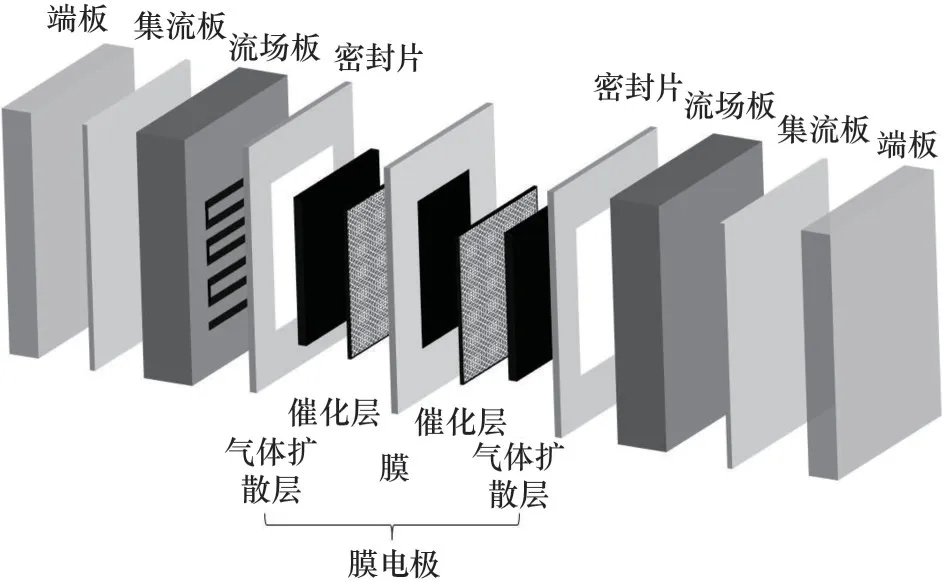
图6 单电池结构示意图Fig.6 The schematic diagram of single-cell structure
4 结语与展望
氨是一种富氢无碳能源载体,具有能量密度高、易液化储存等优势,是理想的储氢介质。将氨直接作为燃料,在低温碱性燃料电池中通过AOR 实现化学能到电能的转化,是氨能源高效利用的理想路径之一。尽管研究者们在氨氧化催化剂的设计制备和NH3-AEMFC 性能优化上已经取得了一定的进展,然而如何加深该研究方向的深入探索和氨燃料电池的商业化应用仍有不少问题需要解决。如图7所示,围绕催化剂设计、构效关系探究和性能优化等方面,比较突出的科学问题有:(1)金属Pt 目前是最为有效的氨氧化催化剂,如何在理论指导下设计高性能低Pt 催化剂依然是研究的重点;非贵金属催化剂的氨氧化活性远低于Pt 基催化剂,如何切实提高非贵金属催化剂的催化性能是该研究方向的难点。(2)尽管当前已有不少报道探究了反应机理及催化剂的作用机制,但在活性中心与氨氧化中的性能强化作用机制依然不够清晰,尤其是在本征活性探索和反应速率方面,需进一步结合实验与理论计算,探究低能垒反应活化路径,用于降低过电位和减少氮氧物种的产生。(3)催化剂的稳定性依然是一个亟待解决的问题,包括粒子团聚、活性物种变化和催化剂中毒等。(4)相比于阳极氨氧化反应,全电池NH3-AEMFC 中存在复杂的界面反应过程,诸多在AOR 中性能优异的催化剂往往在单电池性能测试中表现不佳,需要进一步摸索实际单电池体系下的催化剂作用,这将为NH3-AEMFC 的规模化应用奠定基础。
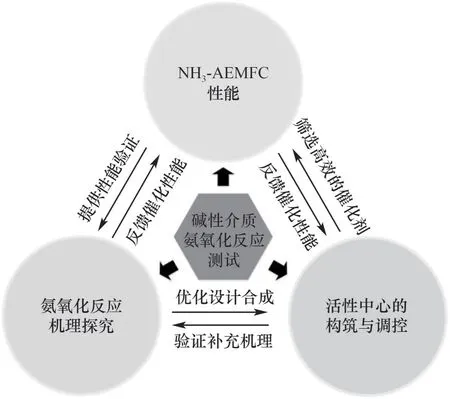
图7 对AOR催化剂设计研究的示意图Fig.7 Schematic diagram of AOR catalyst design study
针对AOR 动力学过程缓慢和催化剂中毒的问题,研究者们进行了大量的研究工作。以Pt 为主的贵金属催化剂,主要研究包含择优取向Pt(100)晶面结构构筑、Pt 纳米颗粒形貌控制、助剂氧化物修饰、载体分散以及合金化等,从而降低贵金属的使用量并减轻催化剂中毒现象,进而提升AOR 活性。在非贵金属催化剂方面,Ni 基催化剂成本低,且起始电位与Pt 相似,但由于联氨(*N2H4)受高能垒限制以及氮氧化物的形成,活性低于Pt 基催化剂。为了提高Ni基催化剂的活性,主要通过控制Ni纳米颗粒的表面结构或与其他金属元素进行合金化来提升活性。然而对于镍基催化剂,无论是反应机理的理解还是催化性能的优化都有待进一步提升。如何设计制备高效稳定的非贵金属催化剂用于NH3-AEMFC 依然是该领域的重点及难点。面对上述的技术难点和突出的科学问题,本文对该方向未来的研究提出了如下建议:(1)将理论计算与实验分析相结合,通过理论计算针对性地对Pt 族贵金属和Ni 基等非贵金属催化剂的结构、组成、合金化等进行分析,获得可行的催化剂设计方案,制备高性能低成本的AOR催化剂;(2)采用原位或在线表征技术(SERS、ATRIR、DEMS 等)检测中间物质,探究在实际反应体系中可能存在竞争或共存关系的反应路径,并结合DFT 理论计算AOR 活性与金属对N 的结合能,为进一步揭示反应过程提供实验理论依据;(3)基于实验数据的分析,借助机器学习手段探究AOR 机理、活性位点以及低能垒反应活化路径,融合合金化、纳米颗粒结构控制以及载体相互作用等多种优势,扩宽电化学反应电势窗口,提高催化剂性能;(4)NH3-AEMFC是多尺度和复杂的界面反应过程,利用实验测试数据和多物理场仿真软件(COMSOL 等)相结合,相互论证并指导MEA 工艺、电池流场和核心组件的优化与开发。
综上所述,低温直接氨燃料电池相对氢燃料电池而言,其性能较低,稳定性不足,处于早期的研究阶段,离实际应用尚有一段距离。作为其补充,氨经热分解转化制氢,然后经质子膜燃料电池发电的间接氨燃料电池在现有阶段具有一定的优势。得益于质子交换膜的成熟工艺,质子膜燃料电池已经实现产品商业化,间接氨燃料电池路线可耦合前端氨分解制氢装置、氢纯化装置,经质子膜燃料电池的系统优化实现产品的场景应用。值得注意的是,相比于质子膜燃料电池的工作温度,间接氨燃料电池路线中氨分解制氢通常在较高温度下工作,在与质子膜燃料电池的耦合过程中,对系统的换热和能效管理要求较高,加之氨制氢装置和氢气纯化装置的存在,间接氨燃料电池系统体积相对较大,更适用于数据中心、基站和大型船舶等固定式电源的应用场景。而对于低温直接氨燃料电池路线,可通过AEMFC直接实现氨能到电能的转化,解决氢的储运难题和安全问题,产品体积小、便携,可面向移动端和小型化的应用场景。因此,间接氨燃料电池路线和直接氨燃料电池路线可以实现应用场景和关键技术路线的优势互补。针对于低温直接氨燃料电池,高效稳定的氨氧化阳极催化剂的设计开发是NH3-AEMFC 性能发展的关键。同时,作为NH3-AEMFC 的核心部分,阴离子聚合物膜、膜电极的制备工艺、双极板的流道设计等也是大幅提升NH3-AEMFC 性能的技术难题。例如,Yan 等[107]已报道集成75 W 的直接氨燃料电池。随着这些关键技术难题的突破,未来直接氨燃料电池路线在清洁能源方面具有广阔前景和应用市场。
猜你喜欢
石油和化工设备(2022年9期)2022-10-18
装备环境工程(2022年8期)2022-09-07
汽车实用技术(2022年10期)2022-06-09
汽车实用技术(2022年5期)2022-04-02
汽车工程师(2021年12期)2022-01-17
当代陕西(2020年23期)2021-01-07
中国计算机报(2020年28期)2020-08-10
智富时代(2018年8期)2018-09-28
智富时代(2018年8期)2018-09-28
智富时代(2018年7期)2018-09-03
