
中空纤维Au-CeZr/FAU催化膜的制备及在富氢气氛CO选择性氧化反应中的应用
2022-10-14 09:40魏李娜彭莉朱锋顾鹏飞顾学红
高等学校化学学报 2022年10期
魏李娜,彭莉,朱锋,顾鹏飞,顾学红
(南京工业大学化工学院,材料化学工程国家重点实验室,南京 211816)
质子交换膜燃料电池(Proton exchange membrane fuel cell,PEMFC)具有操作温度低、启动速度快、使用寿命长、功率密度及能量密度高等特点,是最高效的氢能源利用技术之一.PEMFC以氢气为原料,原料气中含有微量CO.CO极易吸附在电极上,导致电极中毒失活,因此必须消除原料气中的CO,使其浓度低于0.001%[1].CO选择性氧化反应(Carbon monoxide preferential oxidation,CO-PROX)被认为是最经济、有效的去除富H2气氛中CO的方法之一.
自Haruta等[2]发现Au/α-Fe2O3在富H2气氛下优异的低温CO氧化活性和选择性以来,负载型Au催化剂被广泛研究用于CO-PROX.负载型金催化剂的活性受制备方法、金物种的大小及分散度和载体性质等因素的影响[3~6].Zhang等[5]将CeO2负载的纳米Au催化剂用于CO-PROX反应,发现纳米Au催化剂对H2的氧化反应活性低,对CO-PROX反应有较高的反应活性、选择性和稳定性,在70~120℃的温度范围内,CO转化率>99.5%.分子筛具有良好的热稳定性、高的表面积和独特的笼状结构,被广泛用作纳米Au催化剂的载体.Wan等[7]通过离子交换法制备出高活性的低温Au/FAU分子筛催化剂,在30℃时CO转化率高达100%,但反应400 min后催化剂逐渐失活,反应稳定性差.为了进一步提高催化剂的催化活性和稳定性,通常采用合适的金属助剂对载体进行改性.采用离子交换或者浸渍的办法可以很容易地对分子筛进行杂原子改性.Wan等[8]在Fe改性的FAU沸石上制备了Au催化剂并用于COPROX,发现Fe的引入可以有效调节分子筛表面的酸性位,使得负载的Au催化剂表现出更高的反应活性和稳定性.Qi等[9]制备了掺杂Ce的MOR分子筛负载型Au催化剂,发现Ce的引入有利于促进氧化还原平衡的移动,形成更多的Au活性位点.
与颗粒状催化剂相比,催化膜具有比表面积高、化学稳定性好、接触时间可控等优点[7].我们[10]曾将纳米Au粒子负载在中空纤维ZrO2膜表面制备了Au/ZrO2催化膜,并将其用于CO-PROX反应,发现与颗粒催化剂填充的固定床反应相比,反应物与Au活性位的接触更充分,表现出更好的催化活性.我们还采用Zr掺杂FAU分子筛膜负载纳米Au粒子,制备了Au-Zr/FAU催化膜[11],发现Zr在分子筛骨架上的掺杂改善了纳米Au粒子在催化膜表面的分散,使催化膜的活性提高的同时,稳定性也大幅提升.
为了进一步提高催化膜低温下的催化活性,本文采用浸渍的方法将Ce和Zr同时引入分子筛骨架,制备不同Ce/Zr掺杂比例的FAU分子筛膜,然后进一步负载纳米Au,制备Au-CeZr/FAU催化膜,并用于CO-PROX反应,考察了Ce/Zr掺杂比对催化膜结构及性能的影响.此外,还进行了长期催化循环稳定性测试,以评估这种催化膜在实际应用中的可行性.
1 实验部分
1.1 试剂与仪器
六水合硝酸铈[Ce(NO3)3·6H2O]、五水合硝酸锆[(Zr(NO3)4·5H2O]、碳酸钠和氨水均为分析纯,国药集团化学试剂有限公司;四水合氯金酸(HAuCl4·4H2O),分析纯,上海试剂一厂;氢氧化钠,分析纯,西陇化工股份有限公司;偏铝酸钠和硅酸钠均为分析纯,Sigma-Aldrich;无水乙醇,色谱纯,上海凌峰化学试剂有限公司.
MiniFlex 600型X射线衍射仪(XRD),日本Rigaku公司;S-4800型扫描电子显微镜(SEM),日本Hitachi公司;EMAX型能量散射X射线谱(EDX),日本Horiba公司;Optima 7000DV型电感耦合等离子体发射光谱仪(ICP-OES),美国PerkinElmer公司;Tecnai G2 F30型高分辨透射电子显微镜(HRTEM),美国FEI公司;ESCALAB 250X型X射线光电子能谱仪(XPS),美国Thermo Fisher公司;TP-5080型吸附仪,天津先权科技有限公司.
1.2 实验过程
1.2.1 催化膜的制备α-Al2O3中空纤维载体为实验室自制,其平均孔径约为0.65 μm,孔隙率约为48%[12,13].中空纤维FAU分子筛膜采用二次生长法,在水热条件下制备[14].通过浸渍法制备不同Ce/Zr掺杂比例的CexZr2-x/FAU(x=0.5,1,1.5,2)分子筛膜:称取一定质量的Zr(NO3)4·5H2O与Ce(NO3)3·6H2O溶于去离子水中,使其总浓度为2×10-3mol/L,搅拌使其充分溶解,然后将制备好的中空纤维FAU分子筛膜置于上述溶液中,在80℃水浴中离子交换10 h后,洗涤、烘干,于400℃下焙烧4 h,升/降温速率均为1℃/min.配制浓度为1.2×10-3mol/L的HAuCl4溶液,用1 mol/L的NaOH溶液将其pH值调至6.0,稳定24 h.将制备好的CexZr2-x/FAU膜置于上述溶液中,在80℃水浴中离子交换12 h后,洗涤、烘干得到催化膜,命名为Au-CexZr2-x/FAU.催化膜在反应前进行活化处理,活化条件为在空气中100℃焙烧4 h后,在氢气中300℃焙烧1 h.参照文献[11,15]方法,通过二次生长法在自制的α-Al2O3中空纤维载体上制备Au-Zr/FAU催化膜和Au/FAU催化膜.
1.2.2 分析测试方法 采用X射线衍射仪对中空纤维Au-CeZr/FAU分子筛膜的结构及结晶度进行分析.采用扫描电子显微镜观察催化膜表面形貌变化.通过电感耦合等离子体发射光谱仪测定样品元素组成.采用高分辨透射电子显微镜表征催化膜中Au,Ce,Zr元素的微观分布情况,利用均值法测算Au纳米颗粒的平均粒径以及粒径分布,通过量取HRTEM图像上超过100个Au颗粒的粒径进行测算.通过X射线光电子能谱测试催化膜中各元素的价态,使用位于284.8 eV的C1s信号校正结合能数据.利用吸附仪测试H2-TPR和O2-TPD.以FAU分子筛颗粒作为载体,使用与Au-CexZr2-x/FAU催化膜制备过程中相同的离子交换条件制备Au-CexZr2-x/FAU颗粒催化剂,对其进行氧化还原性能表征.
1.2.3 CO选择性氧化反应催化性能测试 通过CO选择性氧化反应测试中空纤维Au-CeZr/FAU分子筛催化膜的反应活性.进入反应器的原料混合气摩尔比为n(CO)/n(H2)/n(O2)/n(He)=0.67∶32.67∶1.33∶65.33,总进料流量为75 mL/min.在25~120℃温度区间内进行反应性能测试,经过催化反应后的混合气体采用安装有TCD检测器的安捷伦GC 7820A气相色谱仪进行在线分析.每个数据点重复测试3次,以获得更可靠的结果.
CO转化率的计算公式如下:

O2的选择性定义为与CO反应的O2与总消耗O2的比:

式中:是进料气中CO的摩尔流量(mol/s);FOutletCO是反应产物中CO的摩尔流量(mol/s);是进料气中O2的摩尔流量(mol/s);FOutletO2是出口流中O2的摩尔流量(mol/s).
2 结果与讨论
2.1 Ce/Zr前驱体溶液浓度配比对催化膜结构的影响
图1(A)为各样品的XRD谱图.从图中可以看出,浸渍法制备得到的Au-CexZr2-x/FAU膜的分子筛骨架结构在制备过程中没有发生坍塌.图1(B)显示的是图1(A)中蓝色部分(2θ=5°~7.5°)的局部放大谱图,为FAU型分子筛(111)晶面特征峰区域.图中并没有出现ZrO2(PDF#37-1484)、CeO2(PDF#43-1002)或Au(PDF#65-2870)的特征峰,说明Au在其中分散均匀,颗粒尺寸较小,Zr和Ce元素没有形成氧化物,而是以离子形式存在于FAU分子筛膜中.值得注意的是,随着Ce掺杂量的增加,FAU的特征峰逐渐向低角度偏移.这是由于Zr4+,Ce3+或Ce4+置换了分子筛骨架中Al的位置,而Zr4+(0.08 nm),Ce3+(0.103 nm)或Ce4+(0.092 nm)的离子半径大于Al3+的离子半径(0.05 nm),因此导致晶面间距增大,衍射峰位向低角度移动[11,16,17].这个结果初步证明了Zr、Ce离子在分子筛骨架中的存在情况.

Fig.1 XRD patterns of Al2O3 support,pure FAU zeolite membrane and Au-CexZr2-x/FAU catalytic membranes prepared with different Ce/Zr precursor solution concentration ratios(A)and partial magnification of the blue area in(A)(B)
Au-CexZr2-x/FAU膜中的Au,Zr,Ce元素含量由ICP-OES测试得到,结果列于表1.可见,随着前驱体溶液中Ce(NO3)3浓度的增大,催化膜中Ce元素的含量增加,Zr元素的含量逐渐减少.

Table 1 Effect of Zr(NO3)4 and Ce(NO3)3 concentrations on metal contents in Au-CeZr/FAU catalytic membranes
图2为Al2O3中空纤维载体、FAU分子筛膜和Au-CexZr2-x/FAU催化膜的SEM照片.Au-CexZr2-x/FAU分子筛膜由于经过浸渍溶液的处理,导致FAU分子筛膜表面出现絮状物质.但XRD表征结果显示FAU分子筛骨架结构仍基本保持完整.选取其中的Au-Ce1Zr1/FAU样品,将膜层刮下一部分进行HRTEM和EDX面扫以观察其微观形貌,结果如图3所示.Au颗粒均匀分散于分子筛膜载体中,平均粒径为(2.94±0.52)nm;EDX元素分布图显示Au,Zr和Ce元素在催化膜中分散均匀.

Fig.2 SEM images of Al2O3 support(A,B),pure FAU zeolite membrane(C,D)and Au-Ce0.5Zr1.5/FAU(E),Au-Ce1Zr1/FAU(F),Au-Ce1.5Zr0.5/FAU(G)and Au-Ce2/FAU(H)catalytic membranes

Fig.3 HRTEM images(A—C)and elemental mappings of the selected area(D—H)of Au-Ce1Zr1/FAU catalytic membrane
利用XPS表征分析焙烧活化前后Au-Ce1Zr1/FAU催化膜中各元素的存在状态.Au4f谱图如图4(A)和(B)所示.焙烧活化前样品中Au元素的价态较复杂,84.4和86.7 eV处的特征峰归属于Au4f7/2,分别对应Au0和Au3+[18];88.2和90.3 eV处的特征峰归属于Au4f5/2,分别对应Au0和Au3+[19].而经过焙烧活化后,属于Au3+的特征峰消失,仅出现Au0的特征峰.金属态Au为CO选择性氧化反应中的活性组分.图4(C)和(D)显示焙烧活化前后归属于Zr3d的峰分别位于182.4和184.8 eV,其中前者为Zr3d5/2轨道结合能,正好处于ZrO(2183.5 eV)[20]和金属态Zr(179.0 eV)[21]的结合能之间,说明Zr元素以Zr4+离子的状态存在于Au-Ce1Zr1/FAU催化膜中.图4(E)和(F)为Ce3d的谱图,标注为V的峰归属于Ce3d5/2能级,标注为U的峰归属于Ce3d3/2能级.其中活化前的样品中,V,V″,V‴和U,U″,U‴的峰对应于Ce4+,V′和U′的峰对应于Ce3+[22,23];活化后的样品中,V′和U′的峰对应于Ce4+,V′和U′的峰对应于Ce3+.Ce4+部分还原为Ce3+,Ce3+所占比例[Ce3+(/Ce3++Ce4+)]由33.6%上升至41.4%.图4(G)和(H)为O1s的谱图,其可以分解为2个峰.在活化前和活化后的样品中,531.8和531.4 eV处的峰Oα归属于表面氧空位或表面活性氧成分O(-2ad)和O(-ad)[24],533.1和532.9 eV处的峰Oβ归属于表面—OH官能团;焙烧后的样品中,O1s的拟合峰整体由532.3 eV移至531.5 eV,向低能级偏移了0.8 eV,Oα所占比例[Oα(/Oα+Oβ)]由57.4%上升至86.0%.这与Ce3d峰中Ce3+(/Ce3++Ce4+)的变化规律相同,可能是由于样品在活化后形成更多的Ce3+,增加了氧空位与表面活性氧所占的比例,因为氧空位和活性氧的生成与相邻的Ce4+/Ce3+离子间还原过程密切相关[25].

Fig.4 XPS analysis of Au-Ce1Zr1/FAU catalytic membrane before and after calcination activation
2.2 氧化还原性能
图5(A)为不同Ce/Zr前驱体溶液浓度配比下制备的Au-CexZr2-x/FAU催化剂颗粒的H2-TPR测试结果.在190℃附近的还原峰对应于Au3+→Au0的还原过程[26,27],这与XPS表征中活化后的样品仅含有金属态的金的结果一致.350℃附近的还原峰对应于表面活性氧物种Ce4+-O-Ce4+[28,29],540℃附近的还原峰对应于Ce4+→Ce3+的还原过程[30,31].Au-Ce0.5Zr1.5/FAU样品仅出现了Au离子的还原峰和比较微弱的表面活性氧物种还原峰,并未出现Ce离子的还原峰,这与该样品中Ce含量较低有关.其它3个样品均出现Au、表面活性氧和Ce的还原峰.其中,Au-Ce1Zr1/FAU样品中Au的还原峰出现温度最低,且强度最大;随着Ce掺杂量的继续增加,Au还原峰位置逐渐向高温区偏移,强度逐渐减弱;表面活性氧的还原峰位置同样随着Ce掺杂量的上升逐渐向高温区偏移,强度略有增强;Ce的还原峰位置无明显变化,强度随着Ce的掺杂量升高而增强.以上H2-TPR结果表明,1∶1的Ce/Zr掺杂比例更有利于Au的分散与还原.
图5(B)为Au-CexZr2-x/FAU催化剂颗粒的O2-TPD谱图.所测试样品的O2-TPD结果中存在3种脱附峰,分别位于250,450和560℃.200~600℃范围的氧脱附峰归属于表面吸附的O-2和O-物种,晶格氧的脱附峰位于600℃以上[24],在测试的样品中并未出现.在Au-Ce1Zr1/FAU中,低温区的氧脱附峰起始温度最低,且强度最高,表明Au-Ce1Zr1/FAU上的氧物种比其它样品上的氧物种活性更高[32,33],这对催化CO选择性氧化反应非常重要.
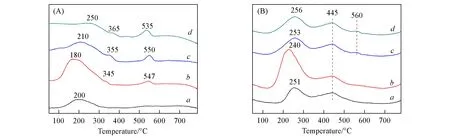
Fig.5 H2-TPR(A)and O2-TPD(B)profiles of Au-CexZr2-x/FAU catalyst particles
2.3 催化反应性能
图6示出了在不同Ce/Zr前驱体溶液浓度配比下制备的Au-CexZr2-x/FAU催化膜、Au-Zr/FAU催化膜以及Au/FAU催化膜的CO选择性氧化反应性能.结合CO转化率和O2选择性可以明显看出,Au-Ce1Zr1/FAU表现出最好的反应活性,40~60℃时CO转化率与O2选择性可以同时达到100%.在不同Ce/Zr掺杂比例的Au-CexZr2-x/FAU催化膜中,催化活性由高到低为Au-Ce1Zr1/FAU>Au-Ce0.5Zr1.5/FAU>Au-Ce1.5Zr0.5/FAU>Au-Ce2/FAU,在60℃下的CO转化率依次为100%,81%,30%和21%,且O2选择性均达到100%.Au-CexZr2-x/FAU催化膜催化性能的变化趋势也表明,随着Ce离子掺杂浓度的增加,催化活性先升高后下降,Ce/Zr离子的比例为1∶1时表现出最佳的催化性能.Wang等[34]通过模型计算发现,Ce/Zr掺杂比为1∶1时活性氧空位的形成能最低,因此具有最高的反应活性.将Au-Ce1Zr1/FAU催化膜催化活性与Au/FAU和Au-Zr/FAU催化膜进行对比,总体上呈现Au-Ce1Zr1/FAU>Au-Zr/FAU>Au/FAU的趋势.仅负载Au的Au/FAU催化膜CO转化率最高仅能达到70%(100℃);相比于Au/FAU催化膜,Au-Zr/FAU催化膜拥有更高的低温CO转化率,在25~60℃时CO转化率为100%,但O2选择性较低,在25~120℃温度范围内均低于Au/FAU催化膜的O2选择性;掺杂了Ce,Zr的Au-Ce1Zr1/FAU催化膜性能进一步提高,在基本保持了Au-Zr/FAU催化膜的高CO转化率的基础上,O2选择性大幅度增强,在40~60℃范围内CO转化率和O2选择性均达到100%.从图中还可以看出,在低温区内,随着温度的升高,各催化膜的催化活性增强,CO转化率提高;而随着温度进一步提高,各催化膜的CO转化率和O2选择性均有不同程度的降低,其原因主要是H2与CO的竞争氧化反应剧烈,使得转化率和O2选择性降低.表2对比了本文制备的Au-Ce1Zr1/FAU催化膜与文献中报道的催化剂的性能,可以看出Au-Ce1Zr1/FAU催化膜的CO转化率和O2选择性均较高.

Fig.6 Catalytic performance in CO-PROX of Au/FAU,Au-Zr/FAU and Au-CexZr2-x/FAU catalytic membranes
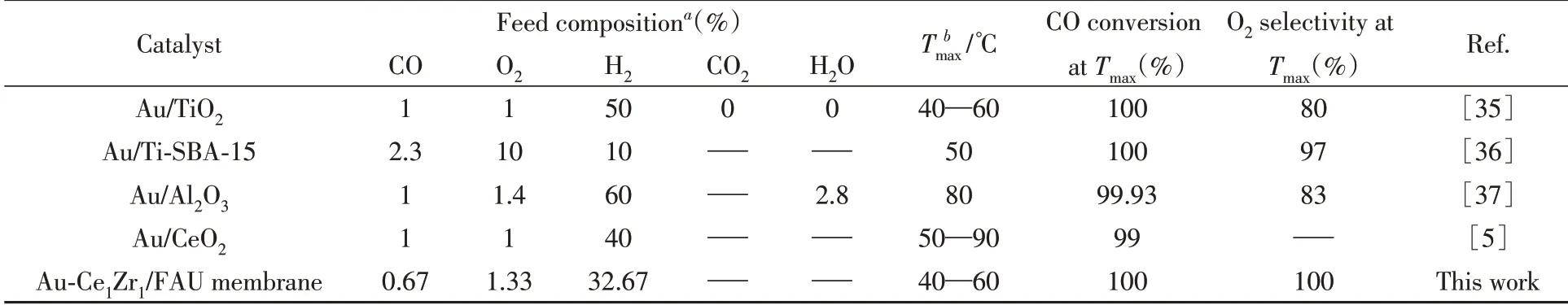
Table 2 Comparison of the catalytic performances of various Au-containing catalysts for CO-PROX reported in the literature
2.4 循环稳定性
实际操作过程通常包含反复的加热和冷却,因此催化膜的热循环性能对于其实际应用十分重要.图7显示了Au-Ce1Zr1/FAU催化膜在25~120℃温度区间的热循环性能.6个测试温度点(25,40,60,80,100和120℃)分别保持100 min以得到稳定的CO-PROX反应结果.可以看出,第一个循环测试区间呈现出最高的CO转化率,反应温度为25℃时CO转化率为70%,40~60℃温度区间内始终保持100%的CO转化率,80℃时CO转化率为96%,120℃时CO转化率最低.随后的10个循环在相同温度点下CO转化率的变化不超过5%,没有明显的性能下降现象,表明Au-Ce1Zr1/FAU催化膜在实际操作过程中具有良好的适应性.

Fig.7 Thermal cycling stability of Au-Ce1Zr1/FAU catalytic membrane between 25 and 120℃
3 结 论
通过浸渍法对FAU型分子筛膜进行Ce、Zr掺杂改性,然后进一步负载纳米Au制备了中空纤维Au-CeZr/FAU催化膜,并研究了其CO-PROX反应性能.系统考察了Ce/Zr掺杂比对催化膜的晶相、表面形貌、元素状态以及氧化还原性能的影响,研究了Au与Ce,Zr间的协同效应对催化膜反应性能的促进作用,比较了不同温度下催化膜的CO选择性氧化催化性能,并对其进行热循环稳定性测试.结果表明,经Ce,Zr离子交换后的FAU分子筛膜保持了完整的晶型结构,Au,Ce,Zr在Au-CeZr/FAU催化膜中分散均匀,当Ce,Zr共掺杂且离子交换液中Ce3+/Zr4+浓度比为1∶1时,更有利于Au的分散与还原,制备得到的Au-Ce1Zr1/FAU催化膜中氧物种活性更高,CO催化氧化反应活性最好,60℃时CO转化率与O2选择性可以同时达到100%,并且经过10次热循环测试后仍然可以保持稳定的催化性能.
猜你喜欢
石油化工(2022年9期)2022-10-19
山西大同大学学报(自然科学版)(2022年3期)2022-07-06
电气技术(2022年5期)2022-05-23
黑龙江大学自然科学学报(2022年1期)2022-03-29
汽车工程师(2021年12期)2022-01-18
第一财经(2019年8期)2019-08-26
作文·初中版(2017年6期)2017-06-16
中学化学(2015年2期)2015-06-05
新课程·中学(2014年7期)2014-10-24
理科考试研究·高中(2014年8期)2014-10-17
