
5-巯基-1,3,4-噻二唑-2-硫酮微溶剂团簇的光谱和理论计算研究
2022-10-14 09:41:00王园月安梭梭郑旭明赵彦英
高等学校化学学报 2022年10期
王园月,安梭梭,郑旭明,赵彦英
(浙江理工大学化学系,杭州 310018)
5-巯基-1,3,4-噻二唑-2-硫酮(MTT)存在MTT(铋试剂I)、2,5-二巯基-1,3,4-噻二唑(铋试剂II,DMT)和1,3,4-噻二唑-2,5-二硫酮(DTT)3种异构体.其衍生物可被用作有机合成原料、分析试剂、燃料添加剂、润滑油添加剂、合成药物中间体、金属的螯合物以及涂料中的闪锈剂和缓蚀剂[1~6].Oyama等[7,8]发现DMT可用作二次锂电池的正极材料.与其它有机硫化合物相比,MTT具有良好的化学稳定性、快速的氧化还原动力学和较低的电荷转移活化能,因此被认为是一种极具潜力的阴极材料[9].MTT和DMT作为金属离子的螯合配体,已广泛应用于金属离子的光度测定[10].2011年,Qin等[11]研究了在酸性溶液中MTT自组装单分子膜(SAM)对铜腐蚀的影响,分子模拟显示,MTT分子通过3个S原子以紧密平行的方式吸附在铜表面并形成亲水性薄膜.
振动光谱已被证明是检测分子间和分子内氢键簇的有效且有价值的方法[12~15].Pope等[16]对MTT及其衍生物的固相进行了红外振动和拉曼散射光谱研究,并且获得了MTT的互变异构体,指认MTT在固相中以硫醇-硫酮式结构稳定存在.直接连接在杂环上的硫代羰基C=S基团的α位为N—H基团,当其受到紫外光照射时,硫酮转化为硫醇[Scheme 1(A)].同样发现,硫脲[17]和硫代乙酰胺[18][Scheme 1(B)]也会发生类似的硫酮→硫醇光异构化.Rostkowska等[19]通过基质隔离技术和理论量子化学计算研究了MTT在紫外光(λ>320 nm)照射下的质子转移反应.结果表明,紫外光能诱导MTT中N—H基团上的氢原子转移到相邻C=S基团的硫原子上形成DMT[Scheme 1(C)].本文通过傅里叶变换拉曼光谱并结合密度泛函理论,表征了固态5-巯基-1,3,4-噻二唑-2-硫酮(MTT)的结构,探究了MTT在不同溶剂及pH条件下的紫外-可见吸收光谱,并结合能量计算确定了MTT在甲醇、水和乙腈中的团簇结构.
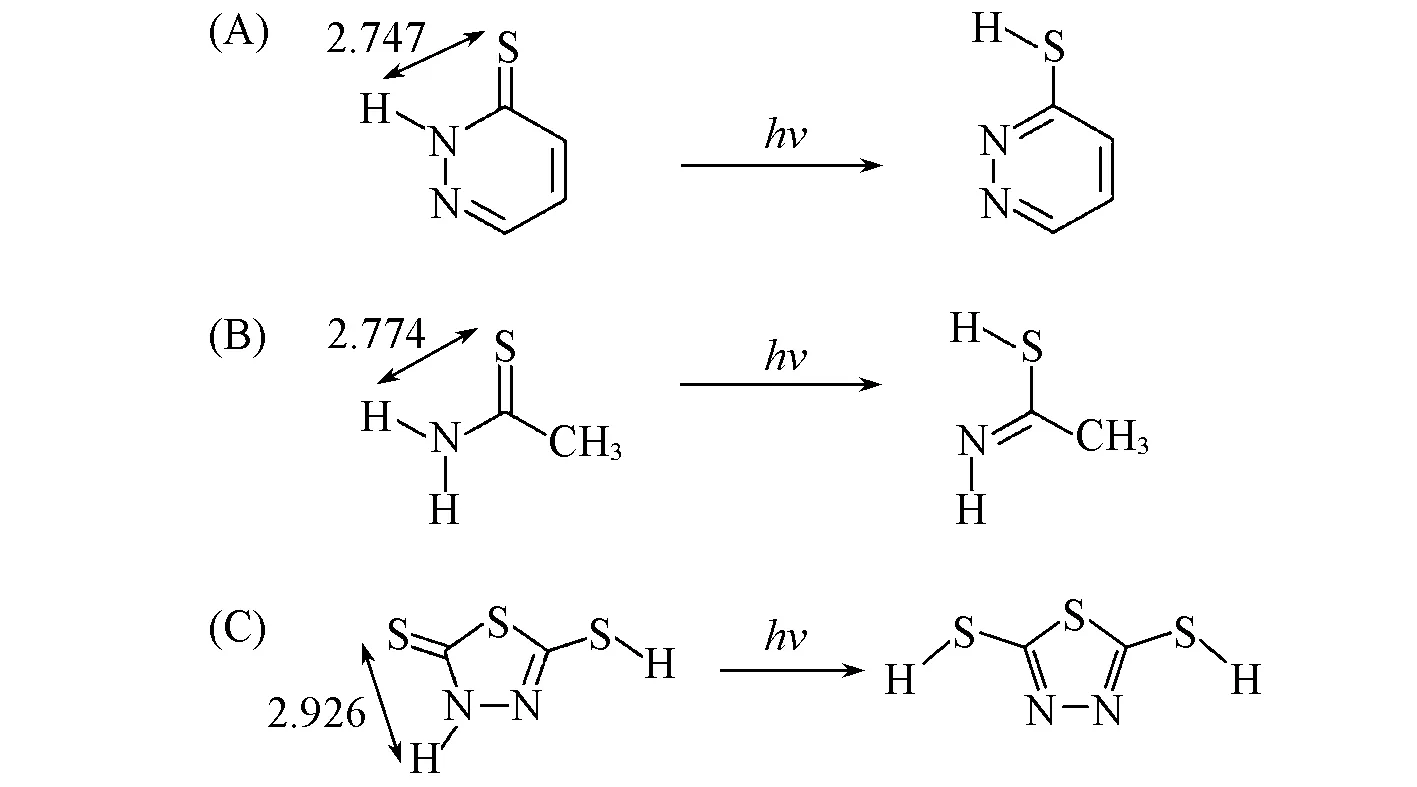
Scheme 1 Ultraviolet light-induced intramolecular proton transfer reactions of six-membered heterocyclic thione and thioamide molecules of 3(2H)-pyridathione(A),thioacetamide(B)and 5-mercapto-1,3,4-thiadiazol-2-thione(C)
1 实验部分
1.1 试剂与仪器
5-巯基-1,3,4-噻唑-2-硫酮(MTT)(纯度98%,北京百灵威科技有限公司);1,3,4-噻二唑-2-硫酮(McT,纯度99%),购于北京百灵威科技有限公司;乙腈和甲醇(纯度99.9%,斯百全化学有限公司);去离子水(自制).
Cary 50 conc型紫外-可见分光光度计(UV-Vis,美国Varian技术中国有限公司);960 Spectrometer型傅里叶变换拉曼光谱仪(FT-Raman,美国Thermo Nieolet公司);PHS-2F型酸度计(上海雷磁公司).
1.2 计算方法
所有计算均采用Gaussian 09[20]程序完成.通过密度泛函理论(DFT)[21]中的B3LYP-D3(BJ)方法和6-311++G(d,p)基组进行了分子团簇的几何构型优化、频率分析和结合能的计算.采用含时密度泛函理论(TD-DFT)计算了优化分子团簇的垂直跃迁能、轨道及振子强度.
2 结果与讨论
2.1 基态结构表征
首先,获得了MTT的FT-Raman光谱.如图1谱线a所示,在1452,658和372 cm-1处出现强振动峰,中等强度的振动峰位于1510,1282,1113,1078,715,535和315 cm-1处,其余振动峰相对较弱.为了归属光谱,在B3LYP-D3(BJ)/6-311++G(d,p)水平下计算了硫醇-硫酮式,硫酮-硫酮式和硫醇-硫醇式3种异构体,其结构和能量如图2所示.从能量来看,MTT的最低,DMT最高,DTT比MTT仅高14.7 kJ/mol.在相同计算水平下的频率分析表明,MTT与实验光谱最吻合,如图1谱线b~d所示.结合Pope等[16]的归属和本文的计算,详细的光谱指认列于表1.将1452 cm-1处的峰归属为N3—C2伸缩与H9—N3—N4面内弯曲的耦合振动,658 cm-1处的峰归属为S1—C5伸缩与S1—C5—N4面内弯曲的耦合振动,372 cm-1处的峰归属为S6—C2伸缩、S7—C5伸缩与C2—N3—N4面内弯曲耦合振动;1510 cm-1处的峰归属为N4—C5伸缩与H9—N3—N4面内弯曲的耦合振动,1282 cm-1处的峰归属为N3—C2伸缩、S6—C2伸缩与H9—N3—N4面内弯曲的耦合振动,1113 cm-1处的峰归属为N3—N4伸缩与C2—N3—N4面内弯曲的耦合振动,1078 cm-1处的峰归属为S1—C5伸缩、S7—C5伸缩、H8—S7—C5面内弯曲、C5—N4—N3面内弯曲与S7—C5—N4面内弯曲的耦合振动,715 cm-1处的峰归属为S6—C2伸缩、S1—C5伸缩与C5—N4—N3面内弯曲的耦合振动,535 cm-1处的峰归属为S6—C2伸缩、S1—C5伸缩、S7—C5伸缩、C2—N3—N4面内弯曲与S1—C5—N4面内弯曲的耦合振动,315 cm-1处的峰归属为S6—C2—S1面内弯曲、S1—C5—N4面内弯曲与S7—C5—N4面内弯曲的耦合振动.
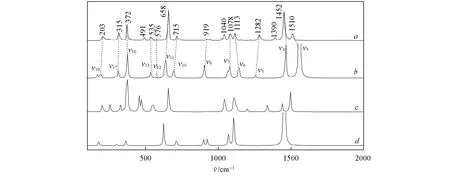
Fig.1 FT-Raman(a)and Raman spectra of MTT(b),DTT(c)and DMT(d)calculated at B3LYP-D3(BJ)/6-311++G(d,p)level

Fig.2 Structures and zero point energies relative to thione-thiol MTT,thione-thione DTT and thiol-thiol DMT calculated at B3LYP-D3(BJ)/6-311++G(d,p)

Table 1 FT-Raman spectra and IR spectrum intensity,Raman activity and assignments calculated at B3LYP/6-311++G(d,p)level
2.2 紫外-可见吸收光谱
MTT在不同溶剂中的稳态吸收光谱如图3(A)所示.MTT在甲醇、水和乙腈中的摩尔消光系数(ε)分别为1.29×104,1.30×104和1.26×104L·mol-1·cm-1,将其中的280~366 nm范围内的吸收峰归属为A吸收带,245~280 nm范围内的吸收峰归属为B吸收带.在甲醇、水和乙腈中,A吸收带的最大吸收波长分别位于334.5,328.0和320.5 nm,B吸收带的最大吸收波长分别为261.1,259.5和269.3 nm.结果表明,对于A吸收带,虽然甲醇和乙腈的极性相近,但甲醇中的最大吸收波长相对于乙腈中的最大吸收峰红移了14 nm.在质子性溶剂水中,相对于乙腈,水的最大吸收波长红移了7.5 nm,这均说明溶剂的氢键和极性作用会同时对轨道跃迁有贡献[22,23].为了进一步探究MTT氢键位点对吸收光谱的影响,开展了1,3,4-噻二唑-2-硫酮(McT)的光谱研究.如图3(B)所示,McT在水、甲醇和乙腈中A带的最大吸收波长分别位于297.1,305.7和307.8 nm.相对于MTT的A带,McT在溶剂中的位移小.相对于水和甲醛,McT在乙腈中发生了红移,而MTT在乙腈中发生了蓝移,可见噻唑环上的巯基对最大吸收波长的位移影响很大.

Fig.3 Steady UV-Vis absorption spectra and molecular structural formula(inset)of MTT(A)and McT(B)in CH3OH,H2O and CH3CN
2.3 不同溶剂中的团簇表征
采用DFT方法在B3LYP-D3(BJ)/6-311++G**水平下优化了可能的氢键团簇结构.图S1~图S3(见本文支持信息)显示了MTT与乙腈、甲醇和水溶剂分子结合可能形成的团簇结构.可见,MTT与水和甲醇分子形成氢键,甚至与乙腈形成氢键[22,23].结果表明,MTT中的—N—HN—C=S基团与H2O,CH3OH分子键合,>NH基团与CH3CN键合,形成氢键MTT(Solvent)n(Solvent=CH3CN,CH3OH和H2O)团簇.总结合能和平均结合能进一步证实了溶剂分子的个数,如图4所示.它们的总结合能均随溶剂分子的增加而增大.当MTT(CH3CN)n中的n=1,MTT(CH3OH)n和MTT(H2O)n中的n=2时,平均结合能最大,结构见图5.由此,初步确定其在乙腈、甲醇和水中的结构分别为MTT(CH3CN),MTT(CH3OH)2和MTT(H2O)2.
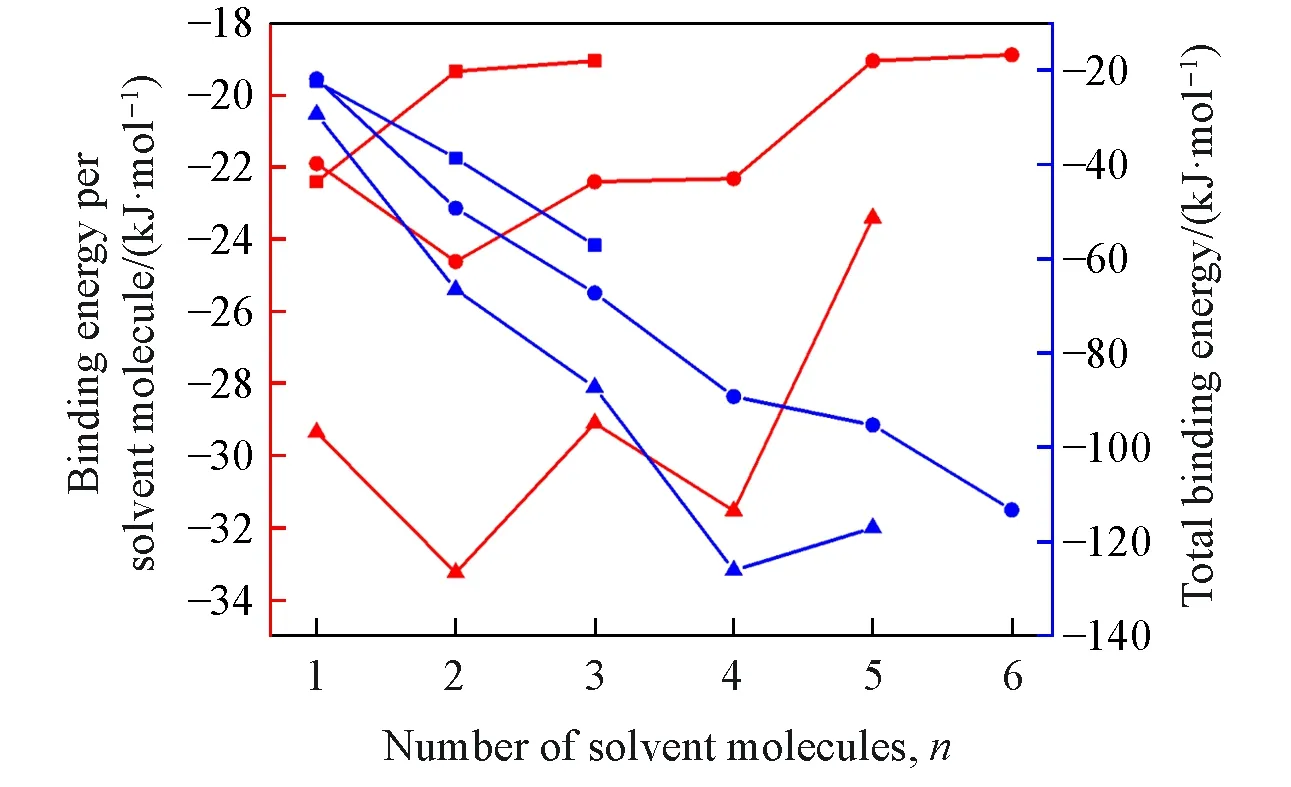
Fig.4 Cluster binding energy of MTT(Solvent)n,and energy per solvent plot at the B3LYP-D3(BJ)/6-311++G**level with polarized continuum model(PCM)solvent model[solvent=CH3CN(■),H2O(●),CH3OH(▲)]
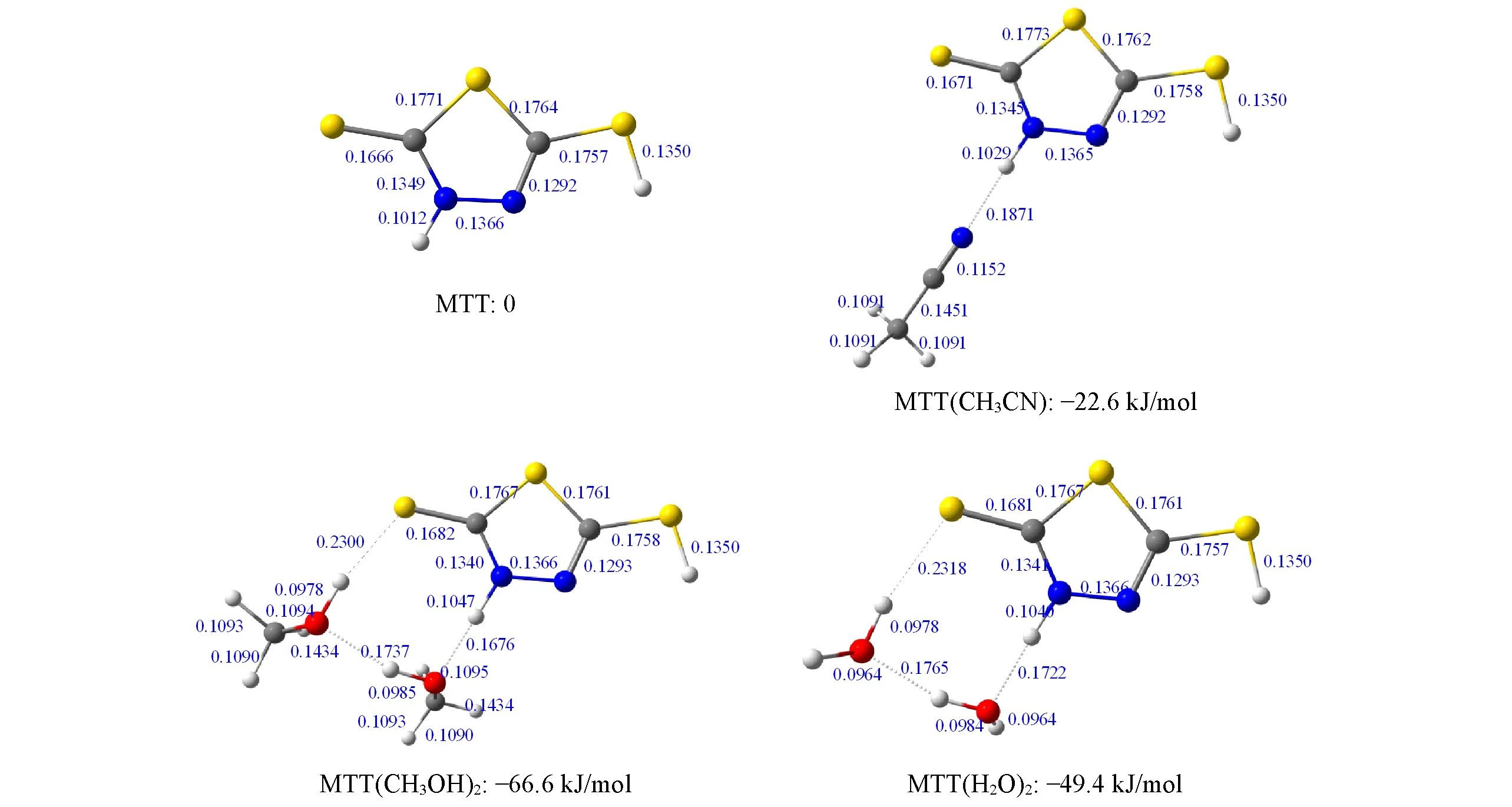
Fig.5 Calculated optimized structures of MTT,MTT(CH3CN),MTT(CH3OH)2 and MTT(H2O)2 cluster,bond length parameter(nm)and zero point energy corrected binding energy at B3LYP-D3(BJ)/6-311++G**(SCRF=PCM)level
如表2所示,在B3LYP/6-311++G**水平上计算了含显性溶剂的MTT团簇在乙腈、甲醇和水溶剂中的最大吸收波长(nm)、电子跃迁类型、电子跃迁能(nm)和振子强度(f).基于上述优化后的MTT(CH3CN),MTT(CH3OH)2和MTT(H2O)2团簇,A吸收带计算的最大吸收带分别为302.9,300.5和299.9 nm,其最大吸收波长对应于第二激发单重态π→π*跃迁(HOMO→LUMO),主要是由C=S的硫原子上的电子部分转移到噻唑环上.计算所涉及的分子轨道如图6(A)~(C)所示.而实验上B吸收带在乙腈、甲醇和水溶剂中的最大吸收带分别为269.3,261.1和259.5 nm,与计算得到的257.4,247.4和246.2 nm位移趋势相同.其中,A吸收带的计算结果与实验的位移趋势有所差异,可能归因于MTT在甲醇和水溶剂的氢键作用对高激发态跃迁的影响更为复杂,仍需进一步的实验与计算验证.此外,若使用隐性溶剂计算得到在乙腈、甲醇和水溶剂中,A吸收带的最大吸收波长分别为300.5,300.3和300.3 nm,B吸收带的最大吸收波长分别为255.3,255.3和254.8 nm,几乎没有明显的位移,如表S1(见本文支持信息)所示.由此可知,与乙腈相比,在甲醇和水中观察到的红移现象主要归因于MTT与溶剂分子之间的氢键作用,可能有更复杂的结构,需要进一步进行振动光谱的指认.

Table 2 Experimental and calculated the maximum absorption(λ),electronic natural transition orbitials(NTOs),transition character and energies and oscillator strength(f)for MTT(CH3CN),MTT(CH3OH)2 and MTT(H2O)2 clusters at(TD)B3LYP(singlet,nstates=15)/6-311++G**level

Fig.6 Molecular orbitals of MTT(CH3CN)(A),MTT(CH3OH)2(B)and MTT(H2O)2(C)clusters at(TD)B3LYP(singlet,nstates=15)/6-311++G**level
进一步探讨了在水和甲醇中pH对MTT的电子吸收光谱的影响.图7分别为MTT在水和甲醇中,随四丁基氢氧化铵的加入而变化的稳态吸收光谱图.由图7(A)可见,在水溶液中,当pH=4.81时,MTT在328 nm处有一个较强的吸收峰.随着碱的加入,在313 nm处的吸收峰逐渐增加,当pH=9.75时不再变化,两者相差15 nm.可推测313 nm处的吸收峰是在基态条件下,MTT在碱性水溶液中去质子化形成阴离子单体,类似于1,2,4-三氮唑-2-硫酮衍生物[24,25].而在甲醇溶液中,MTT随着四丁基氢氧化铵的加入,在400 nm处出现一宽吸收带[图7(B)],由此推测,MTT在碱性甲醇中的反应与水中不同.
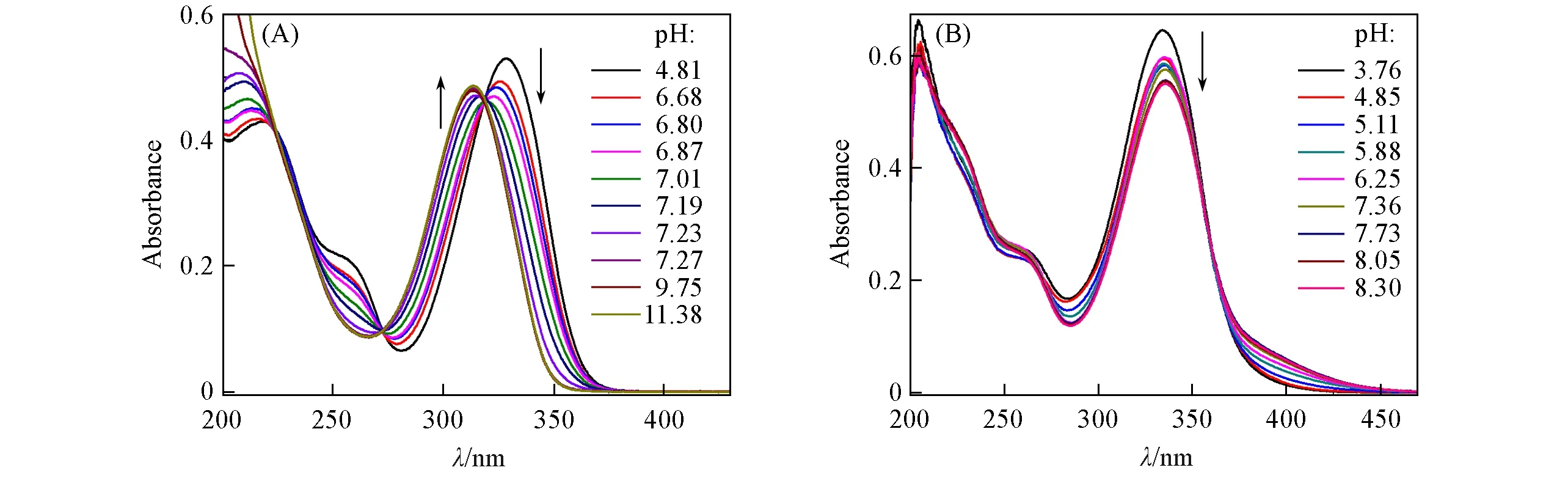
Fig.7 Steady UV-Vis absorption spectra of MTT(5.0×10-5 mol/L)in water(A)and methanol(B)with the addition of tetrabutylammonium hydroxide([(Bu)4N]OH)
3 结 论
通过傅里叶变换拉曼光谱(FT-Raman)与密度泛函理论计算结合表征了固态5-巯基-1,3,4-噻二唑-2-硫酮(MTT)的分子结构.通过与1,3,4-噻二唑-2-硫酮(McT)的紫外-可见吸收光谱的对比表明,MTT分子在溶剂中会与溶剂分子通过氢键作用形成微溶剂团簇,并通过理论计算确认了微溶剂团簇的大小及氢键位点.对不同pH条件下MTT紫外-可见吸收光谱的探究,发现其在碱性水溶液中会去质子化形成阴离子单体.通过优化可能的氢键团簇结构,并结合能量计算初步确定,MTT在乙腈、甲醇和水中的结构分别为MTT(CH3CN),MTT(CH3OH)2和MTT(H2O)2.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20220354.
猜你喜欢
中学生理科应试(2024年1期)2024-05-18 13:02:52
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19 08:38:52
兴义民族师范学院学报(2018年5期)2018-12-18 03:16:54
中国资源综合利用(2017年1期)2018-01-22 02:44:30
山东工业技术(2016年15期)2016-12-01 05:31:08
广州城市职业学院学报(2016年2期)2016-07-25 07:39:30
中国粮油学报(2016年5期)2016-01-23 02:44:53
郑州大学学报(理学版)(2014年4期)2014-03-01 04:21:19
郑州大学学报(理学版)(2014年3期)2014-03-01 04:21:05
原子与分子物理学报(2014年3期)2014-02-28 22:18:23
