
RAFT水溶液聚合制备调驱用凝胶分散体及其性能评价*
2022-10-11 14:33:50杨子腾张丰润泽张艺夕郑憬希刘学敏
油田化学 2022年3期
杨子腾,张丰润泽,张艺夕,郑憬希,刘 灿,刘学敏,张 鹏,伍 波
(1.重庆科技学院化学化工学院,重庆 401331;2.西南石油大学化学化工学院,四川成都 610500;3.中国石油集团渤海钻探工程有限公司油气合作开发分公司,天津 300280;4.四川中泽油田技术服务有限责任公司,四川成都 610000)
随着石油的不断开发,钻采技术不断完善,深部调驱技术得到了广泛的应用[1-3]。深部调驱剂必须满足“进得去、堵得住、可变形、能运移”的特点[4]。常用的调驱剂有预交联凝胶颗粒[5-6]、胶态分散凝胶[7-8]、聚合物微球[9-10]等。其中,预交联凝胶颗粒的制备需要经干燥、粉碎、造粒等工序[11],胶态分散凝胶的制备需借助蠕动泵的剪切作用[12],而聚合物微球则是在有机溶剂存在下经分散聚合或反向微乳液聚合等方法制备[13]。显然,这些调驱剂的制备工艺比较繁琐。自可逆加成-断裂链转移(RAFT)聚合被提出以来[14],RAFT 聚合反应被广泛研究。基于RAFT聚合来制备凝胶分散体,不仅制备工艺简单,而且能满足调驱剂具备微纳米级分子尺度的要求。
龚霞等[15]以N,N′-二乙基二硫代氨基甲酸苄酯为链转移剂,在二甲亚砜溶剂中成功制备了单分散、粒径可达几十纳米的聚丙烯酰胺纳米凝胶。Taton等[16]以乙基黄原酸酯(2-乙氧基硫代硫酰基丙酸甲酯)为链转移剂,在水/醇混合溶液中制备了丙烯酰胺/丙烯酸类聚合物纳米凝胶。本课题组制备的RAFT链转移剂2-(苯甲基三硫代碳酸酯基)丙酸能在有机溶剂中制备微纳米级聚丙烯酰胺凝胶分散体[17]。但该RAFT 链转移剂的水溶性较差,难以实现纯水溶液中的RAFT聚合。上述RAFT链转移剂均为油溶性链转移剂,难以在纯水溶液中进行聚合反应。这一方面限制了其在石油调驱领域的广泛应用,另一方面也存在着可能污染环境的问题。因此,本文首先合成出一种新型水溶性RAFT 链转移剂,并将其应用在纯水相条件下调驱用微纳米级聚丙烯酰胺凝胶分散体的制备中,以期解决传统调驱剂制备工艺复杂以及需要添加有机溶剂的问题,并对其性能进行研究。
1 实验部分
1.1 材料与仪器
硫代乳酸,98%,α-溴苯乙酸,97%,上海麦克林生化科技有限公司;二硫化碳、氢氧化钾、丙烯酰胺(AM)、N,N′-亚甲基双丙烯酰胺(MBA)、过硫酸铵((NH4)2S2O8),分析纯,成都市科隆化学品有限公司;浓盐酸,分析纯,重庆川东化工有限公司。
Agilent 400-MR DD2 型核磁共振仪,美国安捷伦公司;Bruker Tensor 红外光谱仪,德国布鲁克公司;JSM-7800F 扫描电子显微镜,日本电子株式会社;Ntegtra Prima原子力显微镜,俄罗斯NT-MDT公司;MCR102安东帕流变仪,安东帕公司;Brookfield DV2T 黏度计,美国博勒飞公司;Nanophox 纳米粒度分析仪,德国新帕泰克公司;Φ0.55 稀释型乌氏黏度计,台州市椒江玻璃仪器厂。
1.2 实验方法
(1)水溶性RAFT链转移剂的合成
取9.48 g(0.17 mol)氢氧化钾溶于100 mL 蒸馏水中,20 min 之内缓慢加入7.50 mL(0.08 mol)硫代乳酸,再逐滴加入5.07 mL(0.08 mol)二硫化碳后得到橙黄色溶液并不断搅拌,约20 min后加入16.13 g(0.075 mol)α-溴苯乙酸,用三颈烧瓶加热回流10 h后冷却至室温。将溶液置于烧杯中缓慢加入50 mL浓盐酸酸化,根据原料及产物溶解度的不同,采用重结晶的方法进行纯化,真空抽滤后用烘箱干燥5 h可得到水溶性RAFT链转移剂,即S-(2-苯乙酸)-S′-(2-丙酸)三硫代碳酸酯。其合成过程如图1所示。
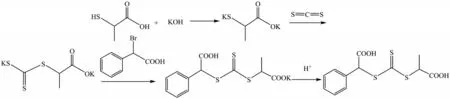
图1 水溶性RAFT链转移剂的合成
(2)聚丙烯酰胺凝胶分散体的制备
向试管中加入去离子水作为溶剂,再按比例加入不同含量的AM、交联剂MBA、合成的水溶性RAFT 链转移剂、引发剂过硫酸铵。溶解完全后向试管中通入氮气5 min 除氧,然后封口。将试管置于一定温度的水浴中聚合。反应完成后,立即取出浸入冰水浴冷却至室温,得到不同尺度的聚丙烯酰胺凝胶分散体。
(3)表征与测试方法
对合成的水溶性RAFT链转移剂用干燥的溴化钾进行压片,然后用红外光谱仪测试。以重水作溶剂,用核磁共振仪对合成的水溶性RAFT 链转移剂进行核磁光谱的表征。
采用扫描电子显微镜(SEM)与原子力显微镜(AFM)分析凝胶分散体的微观形貌及分子尺度。将聚丙烯酰胺凝胶分散体样品稀释并通过22 μm滤网过滤,用纳米粒度分析仪测定凝胶分散体的粒径。
采用黏度计的0号转子测试聚丙烯酰胺凝胶分散体的表观黏度(AV),转速为6~100 r/min。用1 mol/L NaCl 做溶剂,在恒温30 ℃下,用乌氏黏度计测定聚丙烯酰胺凝胶分散体的黏均分子量(MV)。
采用空气轴承同心圆筒CC33.2/PR-XL 转子,用安东帕流变仪测定凝胶分散体的流变曲线;以170 s-1的剪切速率测定凝胶分散体的黏温曲线。以锥板测试系统进行实验,设置应变为10%,选用CP-50号转子进行频率扫描测试来表征凝胶分散体的黏弹性特征。向凝胶分散体中梯度添加氯化钠,用黏度计测定不同矿化度下凝胶分散体的黏度变化。向凝胶分散体中逐渐加入极少量的氢氧化钠调节pH 值,用黏度计测定不同pH 值下凝胶分散体的黏度变化。
2 结果与讨论
2.1 链转移剂的制备与结构表征
合成的水溶性RAFT 链转移剂的红外光谱图(图2)中,3425.98 cm-1处的吸收峰是由于羧酸羟基的强烈缔合作用形成氢键,致使—O—H 伸缩振动形成很宽的吸收带;1720.35 cm-1处为羧酸羰基C=O的伸缩振动峰,两处伸缩振动峰证实了—COOH 的存在。1643.55 cm-1处为苯环的伸缩振动峰,1380.68 cm-1处为甲基的伸缩振动峰,1229.12 cm-1处为碳硫双键的伸缩振动峰,608.50 cm-1为碳硫单键的伸缩振动峰。以上分析初步证实合成的水溶性RAFT链转移剂的结构中含有羧基、苯环、碳硫单键、碳硫双键及甲基等基团或结构。
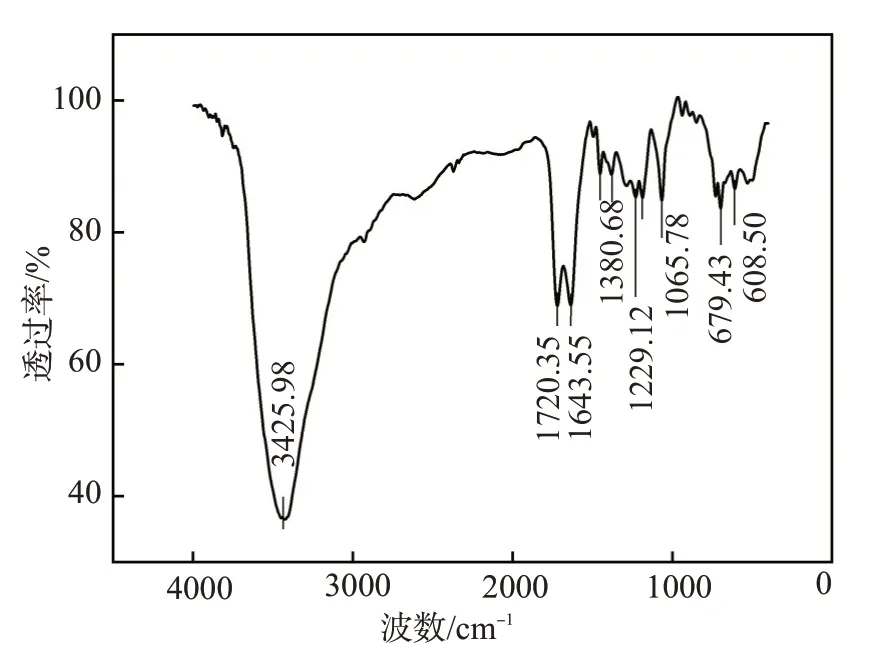
图2 水溶性RAFT链转移剂的红外光谱图
合成的水溶性RAFT链转移剂的核磁共振氢谱图(图3)中,7.407~7.369处的振动峰为苯环上H的化学位移,4.930~4.745 处为与苯环相连的次甲基中H 的化学位移,3.577~3.519 处为与甲基相连的次甲基中H的化学位移,1.482~1.168处为甲基中H的化学位移。5.1 左右处的峰可能是α-溴苯乙酸自身相互作用生成的副产物中H 的化学位移。由于使用了重水作溶剂,发生了质子交换效应,导致羧酸上的活泼H 无法检测。以上结果分析表明产物结构与合成路线中所示结构相一致,即所合成的水溶性RAFT链转移剂为S-(2-苯乙酸)-S′-(2-丙酸)三硫代碳酸酯。
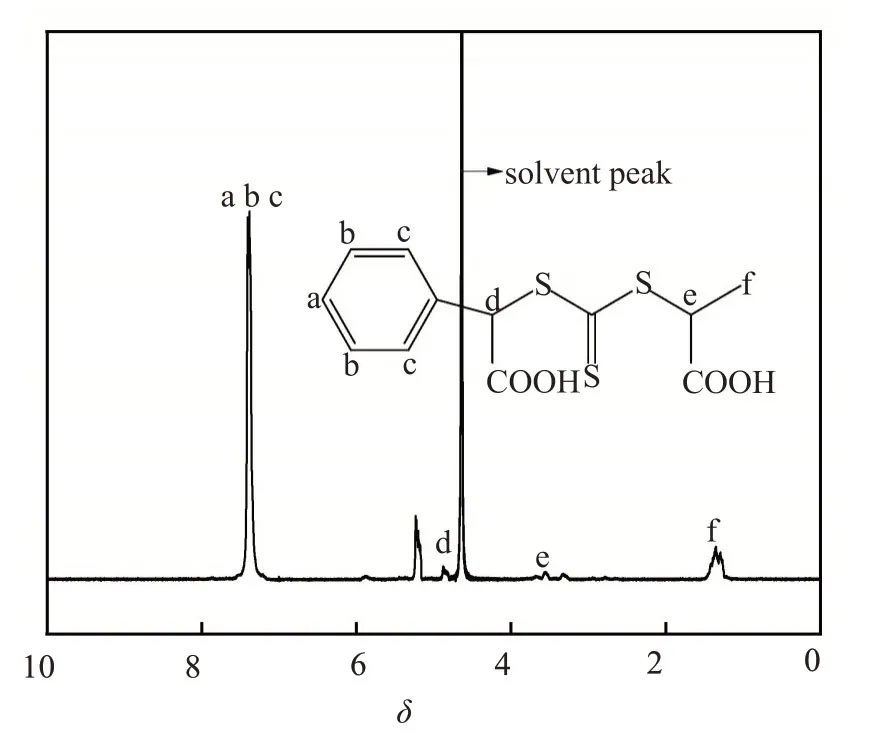
图3 水溶性RAFT链转移剂的核磁共振氢谱
2.2 凝胶分散体的微观形貌及粒径
按表1中序号8的实验条件制备聚丙烯酰胺凝胶分散体。用AFM和SEM观察凝胶分散体的微观形貌,结果如图4所示。RAFT水溶液聚合制备的聚丙烯酰胺凝胶分散体的微观形貌为分散的不规则球状结构。对凝胶分散体的SEM 图进行粒径统计分析,其粒径大小在0.92~6.13 μm之间。由粒径分布图(图5)可见,凝胶分散体的粒径主要分布在1.5~2.5 μm。综合以上分析可得,RAFT 水溶液聚合制备的聚丙烯酰胺凝胶分散体的分子尺度为微米级别。

图4 凝胶分散体的微观形貌
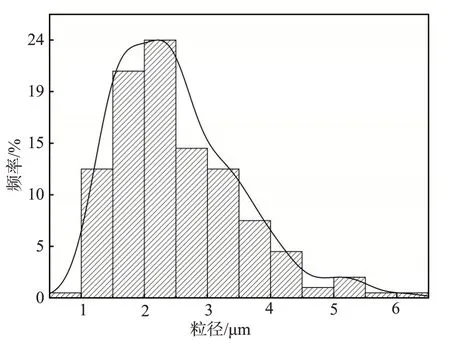
图5 凝胶分散体的粒径分布图
对按表1 中4~8 号实验条件制备的凝胶分散体进行过滤处理后,用纳米粒度分析仪测得其平均粒径依次为55.42、86.76、48.14、75.92、48.43 nm。所制备的凝胶分散体经过滤处理后的粒径范围在48~87 nm 之间,其粒径随链转移剂用量的变化无明显规律,但证实了凝胶分散体中存在纳米尺度级别范围的颗粒。通过以上AFM、SEM 以及纳米粒度分析仪对凝胶分散体微观形貌及粒径的表征,基本确定RAFT水溶液聚合的聚丙烯酰胺凝胶分散体的分子尺度为微纳米级别。
2.3 反应条件对凝胶分散体的影响
2.3.1 反应物配比的影响
在聚丙烯酰胺凝胶分散体的制备中,交联剂可以使聚丙烯酰胺凝胶化,而RAFT 链转移剂可以避免聚丙烯酰胺形成整体凝胶,从而可以制备出凝胶分散体,可见反应物配比对凝胶分散体的性能会有一定影响。反应物配比对凝胶分散体性能的影响如表1 所示。引发剂引发单体生成增长自由基,增长自由基与RAFT 链转移剂中C=S 双键可逆加成,形成不稳定的中间体自由基,进而S—R 键断裂生成“休眠种”以及新的自由基R·,R·又再次引发单体聚合,以此循环。可逆加成-断裂链转移聚合的结果是生成的聚合物分子量分布窄,聚合物链较短、分子量较小。随着RAFT链转移剂与交联剂比值的增大,聚丙烯酰胺由不可流动的凝胶状态逐渐变为可流动的液体状态,即成为凝胶分散体。同时,其黏度不断降低,黏均分子量在总体上也成降低的趋势。链转移剂用量较小时,不足以发挥其链转移效率,而交联剂则起主要作用,即形成整体凝胶。随着链转移剂用量的增多,其中间体自由基的浓度增大,形成了更多的“休眠种”,链转移效率越好。因此,考虑到链转移剂的加量成本,控制单体、交联剂及链转移剂的物质的量比为95.86∶0.1258∶4.0142时,凝胶分散体的性能较好。
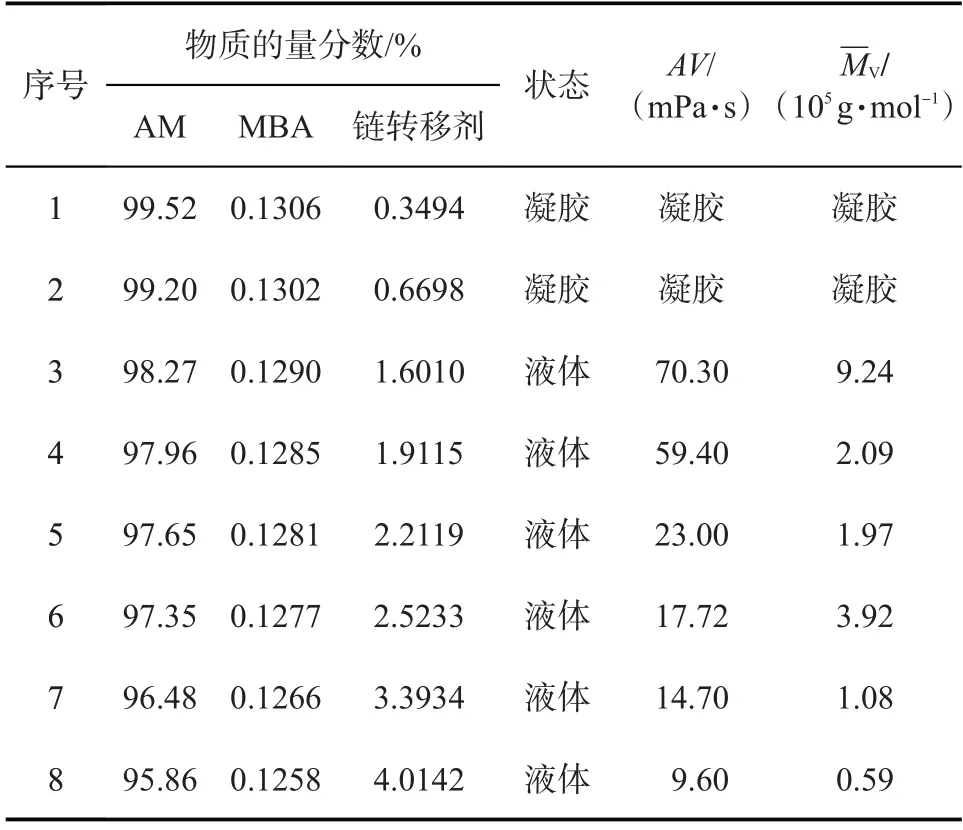
表1 反应物配比对凝胶分散体性能的影响
2.3.2 聚合温度的影响
在聚丙烯酰胺凝胶分散体的合成中,RAFT 聚合采用热引发方式。聚合温度对凝胶分散体性能的影响如表2 所示。在相同反应物物质的量比、聚合时间(2 h)和固含量(6%)下,随着温度的升高,凝胶分散体的黏度和黏均分子量总体上呈降低的趋势。当聚合温度较低时,不足以提供RAFT 链转移剂加成断裂形成中间体自由基所需的能量,导致链转移效率较低。随着聚合温度的升高,链转移作用逐渐增强。聚合温度为70 ℃时,凝胶分散体的性能最佳。
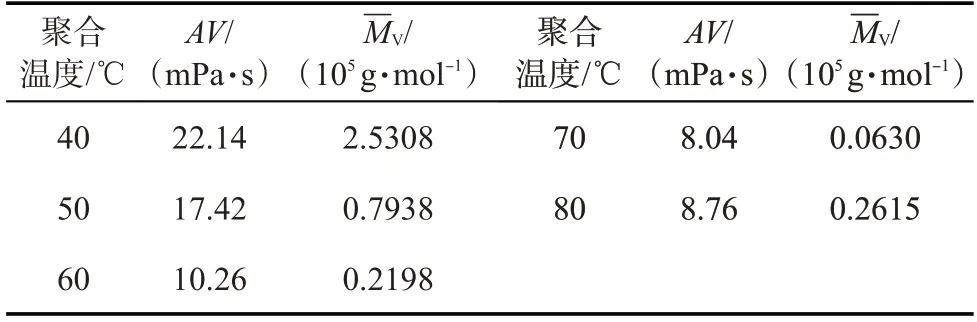
表2 聚合温度对凝胶分散体性能的影响
2.3.3 聚合时间的影响
聚合时间决定了RAFT 聚合反应的进行程度,其对聚丙烯酰胺凝胶分散体性能的影响如表3 所示。在70 ℃下,随着聚合反应的不断进行,凝胶分散体的黏度先降低后增大,黏均分子量在总体上也表现出相应的趋势。当聚合时间较短时,单体自由基浓度较高,而RAFT 链转移剂加成断裂形成的中间体自由基较少,链转移效率低,链终止的几率增大。随着时间的延长,链转移效率逐渐增大,降低了链终止的发生几率,使凝胶分散体的黏度较低。聚合反应再继续进行时,RAFT链转移剂反应完全,此时体系中聚合反应速率大于链转移速率,其黏度和黏均分子量增大。
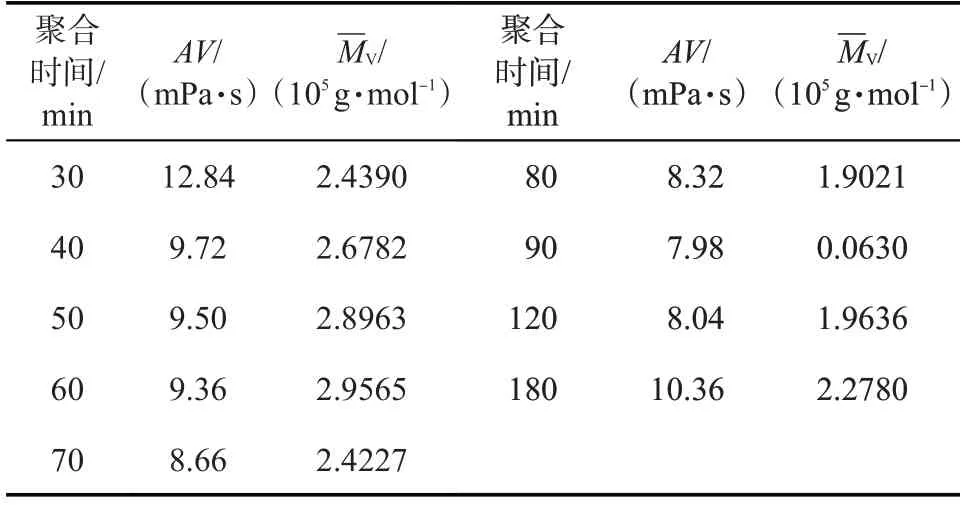
表3 聚合时间对凝胶分散体性能的影响
2.3.4 固含量的影响
固含量反映了体系中的反应物浓度的大小。在聚合温度为70 ℃、聚合时间为2 h的条件下,固含量(质量分数)对聚丙烯酰胺凝胶分散体性能的影响如表4 所示。随着固含量的增加,凝胶分散体的黏度和黏均分子量增加。固含量较高时,即聚合体系中溶剂的量较少,制得的凝胶分散体浓度增大,表现为凝胶分散体的黏度和黏均分子量增大。
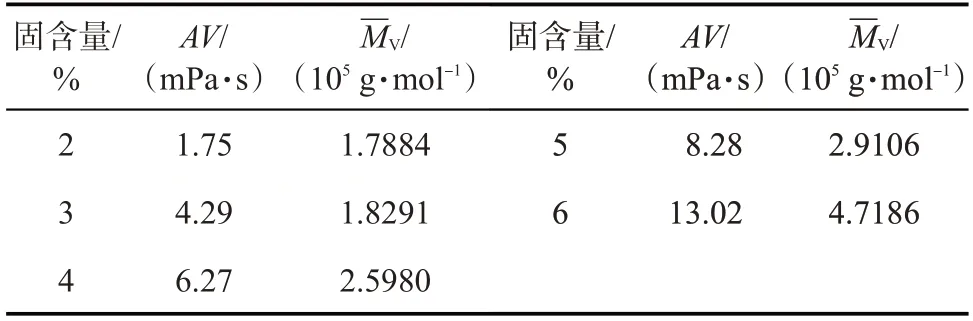
表4 固含量对凝胶分散体性能的影响
通过以上分析,制备聚丙烯酰胺凝胶分散体的最优条件为:单体、交联剂及链转移剂的物质的量比为95.86∶0.1258∶4.0142,引发剂的质量分数为0.05%,固含量不超过6%,聚合温度为70 ℃,聚合时间为2 h。
2.4 凝胶分散体的性能评价
2.4.1 流变性及黏弹性
在AM、MBA、(NH4)2S2O8的物质的量比为220∶0.8∶0.8、固含量为3.5%、链转移剂的物质的量分数分别为2.5%和3.0%的条件下制得聚丙烯酰胺凝胶分散体,用流变仪测试其流变曲线,结果如图6 所示。凝胶分散体属于剪切稀释型流体,在低剪切速率下,凝胶分散体的黏度降低较快;当剪切速率较高时,凝胶分散体的黏度变化较小,趋于牛顿流体的特征。由于凝胶分散体由凝胶单元颗粒构成,在低剪切速率下,颗粒间的摩擦力较强,黏度较高;随着剪切速率的增大,颗粒间的摩擦力迅速降低,最后保持稳定。链转移剂加量越高,制得的凝胶分散体中的凝胶颗粒较小,表现出的黏度越低。
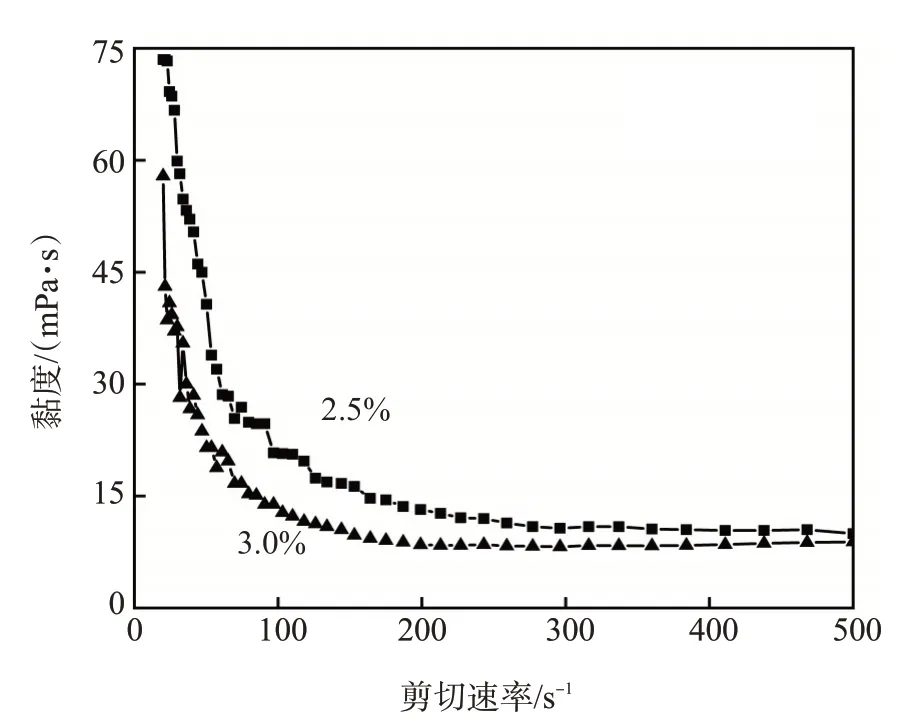
图6 凝胶分散体的流变曲线
凝胶分散体的黏弹性如图7所示。凝胶分散体的耗能模量G′′均高于储能模量G′,G′非常小,即凝胶分散体表现为黏性流体特性,进一步证实了凝胶分散体是由微小的凝胶单元颗粒分散在水溶液中形成的。
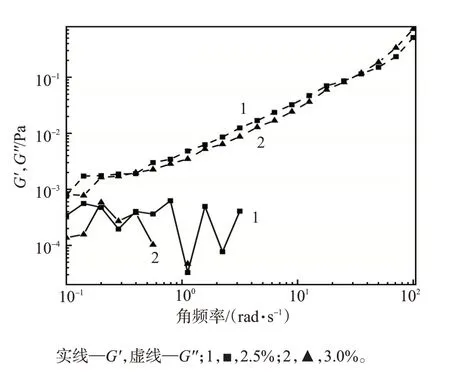
图7 凝胶分散体的黏弹性曲线
2.4.2 温度响应性
温度影响了分子间作用力,进而影响了凝胶分散体的黏度。凝胶分散体的黏度随温度的变化如图8所示。凝胶分散体的黏度随着温度的升高呈现降低的趋势,但降幅较小。当温度逐渐升高,分子间作用力减小,凝胶分散体的黏度也随之降低。同时,温度升高使得凝胶分散体中凝胶单元颗粒之间的黏附效应降低,导致黏度降低。然而,凝胶分散体的初始黏度较小,使得黏度随温度升高而降低的幅度较小。此外,由于链转移剂中含有苯环,即凝胶分散体结构中亦含有苯环,这可能使得凝胶分散体具有一定的耐温能力。
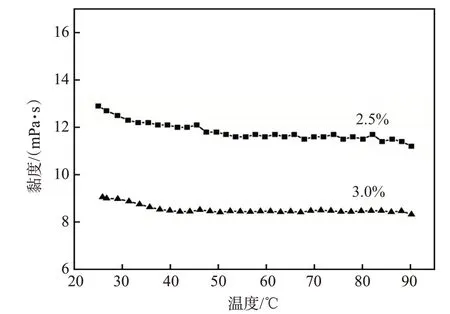
图8 凝胶分散体的黏温曲线
2.4.3 盐度响应性
向聚丙烯酰胺凝胶分散体中梯度加入等量氯化钠,用黏度计测得其盐度响应性曲线如图9 所示。随着矿化度的增加,凝胶分散体的黏度变化不大。RAFT水溶液聚合制备的凝胶分散体受矿化度影响较小。这是由于盐主要通过电荷作用来影响聚合物性质,而凝胶分散体的电荷较弱甚至不带电荷,因而凝胶分散体受矿化度的影响较小。
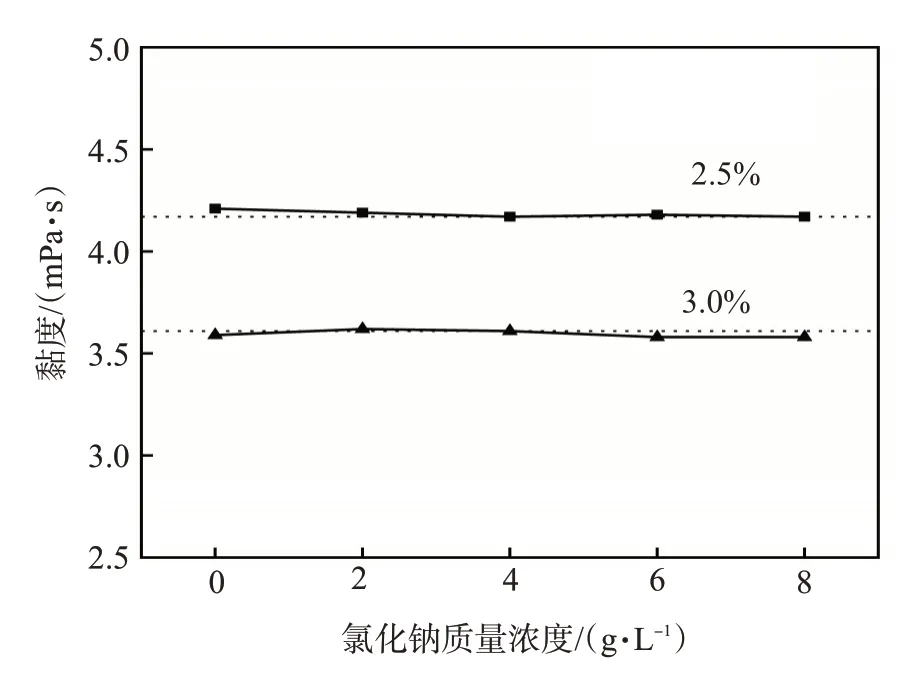
图9 凝胶分散体的盐度响应性曲线
2.4.4 pH响应性
向聚丙烯酰胺凝胶分散体中逐步加入极少量的NaOH来调节pH值。凝胶分散体的pH响应性如图10所示。由于链转移剂结构中含有两个羧基,因而凝胶分散体溶液呈酸性。当加入NaOH后,pH值逐渐增大,凝胶分散体的黏度逐渐降低;当pH值继续增大,其黏度降幅较为明显。这是由于NaOH 与凝胶分散体结构中的羧基反应,形成了离子型凝胶分散体,凝胶颗粒间的斥力增加,降低了凝胶颗粒的聚集,使得凝胶分散体的黏度有所降低。
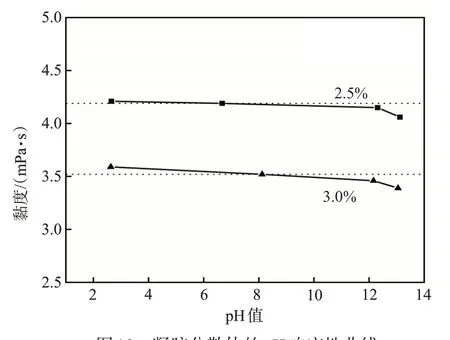
图10 凝胶分散体的pH响应性曲线
3 结论
以硫代乳酸、二硫化碳、α-溴苯乙酸为原料制得水溶性RAFT 链转移剂,并在纯水相条件下制得调驱用微纳米级聚丙烯酰胺凝胶分散体。与传统调驱剂制备工艺相比,制备时间缩短、制备条件简化,无需添加有机溶剂,产物分子尺度小。制备聚丙烯酰胺凝胶分散体的最优条件为单体、交联剂及链转移剂的物质的量比为95.86∶0.1258∶4.0142,引发剂的质量分数为0.05%,固含量不超过6%,聚合温度为70 ℃,聚合时间为2 h。聚丙烯酰胺凝胶分散体接近牛顿流体特性,其黏弹性表现出黏性流体特性,进一步证实了凝胶分散体是由微小的凝胶单元颗粒分散在水溶液中形成的。凝胶分散体的黏度随温度和pH值的升高而降低,但降幅较小,其黏度几乎不受矿化度的影响。在凝胶分散体的制备中,RAFT 链转移剂的加量远低于单体的加量,所增加的RAFT链转移剂成本与凝胶分散体的制备优势和粒径优势相比,仍是可以接受的,这也有望在油田调驱领域得到进一步应用。
猜你喜欢
云南化工(2021年11期)2022-01-12 06:06:18
云南化工(2021年5期)2021-12-21 07:41:26
煤炭与化工(2021年6期)2021-08-06 10:04:10
蔬菜(2016年8期)2016-10-10 06:48:52
安徽大学学报(自然科学版)(2016年2期)2016-09-20 12:09:27
中成药(2016年4期)2016-05-17 06:07:52
现代食品(2016年24期)2016-04-28 08:12:08
当代化工研究(2016年2期)2016-03-20 16:21:20
有机氟工业(2014年4期)2014-06-01 12:30:37
弹性体(2014年1期)2014-05-21 02:05:37
