
Tensile properties of functionalized carbon nanothreads
2022-10-09 07:24:58HifeiZhnJingShngChofengYuntongGu
Namo Materials Science 2022年3期
Hifei Zhn ,Jing Shng ,Chofeng Lü ,Yuntong Gu,c,**
a Department of Civil Engineering,Zhejiang University,Hangzhou,310058,PR China
b School of Mechanical,Medical and Process Engineering,Queensland University of Technology (QUT),Brisbane,QLD,4001,Australia
c Center for Materials Science,Queensland University of Technology (QUT),Brisbane,QLD,4001,Australia
Keywords:Carbon nanothread Functional group Tensile deformation Stress concentration Molecular dynamics simulation
ABSTRACT Low dimensional sp3 carbon nanostructures have attracted increasing attention recently,due to their unique properties and appealing applications.Based on in silico studies,this work exploits the impacts from functional groups on the tensile properties of carbon nanothreads (NTH)– a new sp3 carbon nanostructure.It is found that functional groups will alter the local bond configuration and induce initial stress concentration,which significantly reduces the fracture strain/strength of NTH.Different functional types lead to different local bond reconfigurations,and introduce different impacts on NTH.Further studies reveal that the tensile properties decreases generally when the content of functional groups increases.However,some NTHs with higher content of functional groups exhibit higher fracture strain/strength than their counterparts with lower percentage.Such observations are attributed to the synergetic effects from the sample length,self-oscillation,and distribution of functional groups.Simulations show that the tensile behaviour of NTH with the same functional percentage differs when the distribution pattern varies.Overall,ethyl groups are found to induce larger degradation on the tensile properties of NTH than methyl and phenyl groups.This study provides a comprehensive understanding of the influence from functional groups,which should be beneficial to the engineering applications of NTH.
1.Introduction
Low-dimensional carbon nanostructures are ideal building blocks for a broad range of applications due to their excellent physical and chemical properties[1],such as next generation devices,flexible electronics,and high-performance materials.Carbon possesses three different hybridization states,including sp1,sp2,and sp3bonds,these different types of C bonds as well as their combination enable the existences of many different carbon nanostructures,ranging from zero to three-dimensional(3D).Among them,the sp2carbon nanostructures,including fullerene,carbon nanotube(CNT)and graphene,have received the most intensive interests from academic and engineering communities in past decades.The sp2bonded carbon nanostructures normally have supreme in-plane mechanical properties,for example,carbon nanotube or graphene is reported with a Young's modulus up to 1 TPa [2,3].However,their out-of-plane mechanical properties frequently fail the real engineering requirements.For instance,the thin-wall carbon nanotube(CNT)suffers from lateral flattening (or buckling),and the graphene layer can hardly maintain a flat configuration in experiments [4].These circumstances drive researchers to revisit the typical sp3bonded carbon structure–diamond,which is regarded as the hardest material in nature.Recent experimental works show that a two-dimensional (2D) diamond can be prepared based on bilayer graphene[5,6],which has been predicted with excellent in-plane and out-of-plane mechanical properties[7,8].
Besides 2D diamond,the one-dimensional (1D) sp3bonded nanostructures also attracted great attention [9].Earlier works prepared diamond nanowire or nanorod through reactive-ion etching (RIE) technique [10,11] or chemical vapor deposition (CVD) method [12].They have been shown with good electrochemical properties,excellent electron field emission,mechanical properties [13],and envisioned with appealing applications in biomedical fields.However,the ineffectiveness and challenges of the REI or CVD method have greatly impeded their research.Recently,researchers successfully synthesized an ultra-thin sp3bonded 1D nanostructure,named as carbon nanothread(NTH),through high-pressure (20 GPa) solid-state reaction of benzene at room temperature[14].NTH is reported with a diameter less than 0.5 nm,such small diameter leads to significant challenges for structural characterization[15,16].Despite that,potential configuration of NTHs have been systematically enumerated based on first principles calculations,including the fully saturated (or degree-6) and partially saturated (or degree-4)structures [17,18].Preliminary works report that NTH has tunable mechanical properties[19–21]and thermal transport properties[22,23].Its non-smooth surface is shown to be beneficial for the applications as reinforcements for polymer nanocomposites [24] and carbon nanofibers[25–27],due to the high interfacial load transfer efficiency.
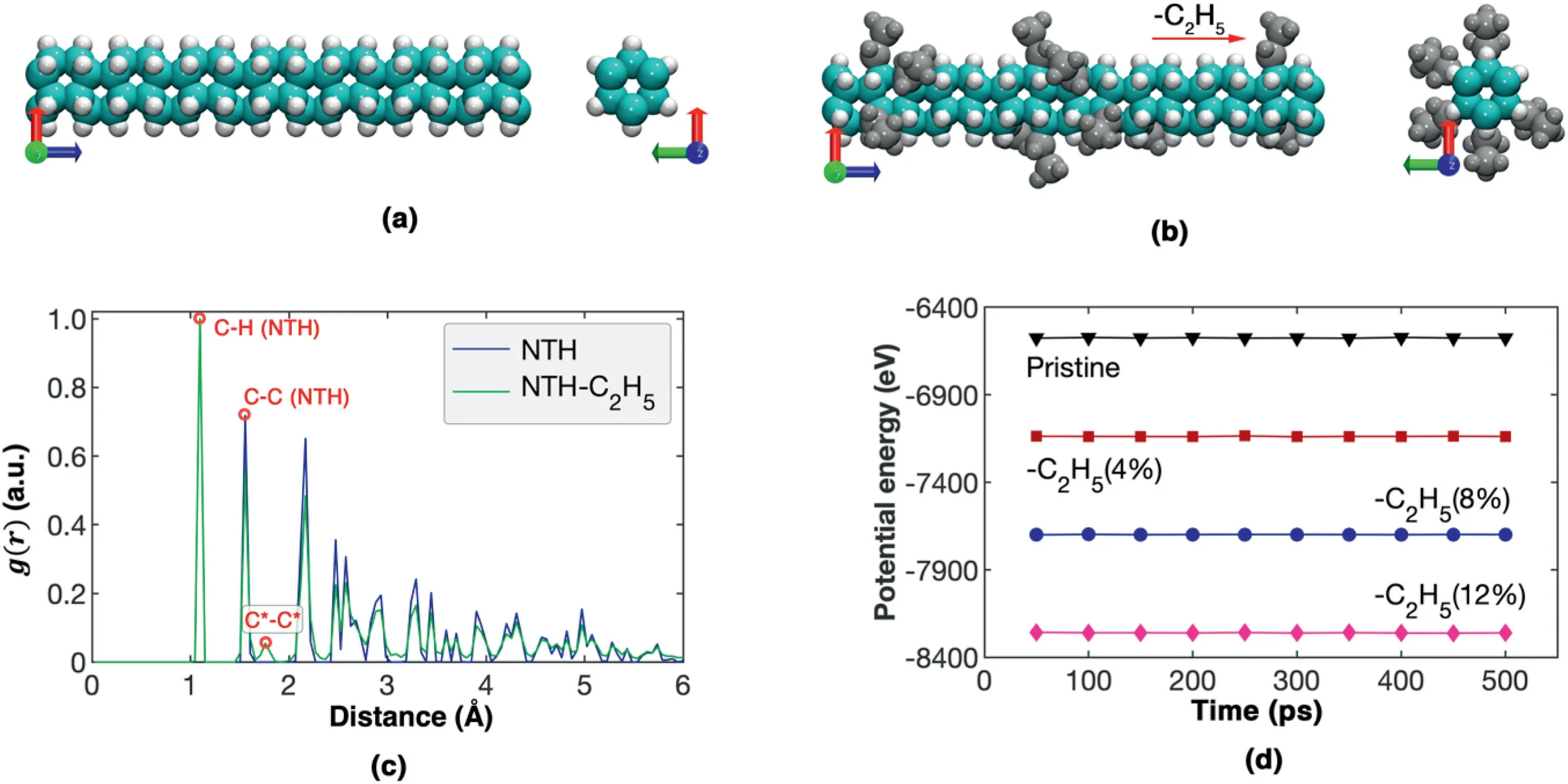
Fig.1.Schematic view of the model.(a)The pristine NTH;(b)The NTH with randomly dispersed ethyl functional groups;(c)Comparisons of the radial distribution function g(r)for NTH with and without ethyl functional groups;(d) The potential energy trajectory during the relaxation for the NTH with different percentages of ethyl functional groups.
Functionalization is one of the effective ways to improve the interfacial load transfer efficiency for low dimensional nanostructures used in composite structures.One of the advantages of NTH over CNT is that NTH possesses a fully hydrogenated surface,which enables a further functionalization without introducing additional defects to the backbone structure.It is commonly reported that functionalization will significantly deteriorate the mechanical properties of CNT due to the C bond change from sp2to sp3.In comparison,no such bond change will occur for the NTH with functionalization,i.e.,the C bonds remain as sp3.Previous density functional theory calculations suggest that the NTH maintain a nearly undegraded mechanical properties with the addition of–CH3,–NH2,–OH,and–F groups[28].However,this work considered a small unit cell with periodically added functional groups,which does not reflect the actual NTH with a finite length and randomly distributed functional groups.To this end,this work aims to answer the question whether the NTH with a finite length can maintain its tensile performance after functionalization.According to the molecular dynamics(MD) simulations,the functional group will induce local bond change,which induces local stress concentration.As a result,although Young's modulus only receives a minor reduction,its fracture strain/strength experiences a significant degradation.
2.Computational methods
As illustrated in Fig.1a,the tube (3,0) NTH was considered in this work,which has a straight tubular structure like the fully hydrogenated(3,0) CNT.Based on experiments [29] and density functional theory calculations[28],NTHs can be modified by different functional groups,such as–CH3,–NH2,–OH,and–F.For example,the–NH2decorated NTH can be prepared by compressing aniline in crystal phase-II [29].In this work,the ethyl group (–CH2CH3) was considered initially,which randomly replaced the surface H atoms of the NTH(Fig.1b).The H atoms to be replaced by functional groups were randomly selected by our in-house code,and the maximum number of functional groups in each benzene ring was three to avoid strong local bond distortion.The functionalization percentage is defined as the ratio between the number of the replaced H atoms (or the number of functional groups) and the total H atoms.The C–C and C–H atomic interactions were described by the commonly used intermolecular reactive empirical bond order (AIREBO) potential,which was established based on different hydrocarbon configurations[30,31].This potential has been shown to well reproduce the thermal and mechanical properties of different carbon nanostructures[32–34].Recent works show that AIREBO potential can well predict the mechanical properties NTHs compared with the density functional theory calculations[35]or other NTHs with similar configuration[36].To avoid the spurious high stress at high bond strain,the cut-off distance of the AIREBO potential was chosen as 2.0 Å [37–39].The NTH with and without ethyl groups share a similar radial distribution function profile(Fig.1c),suggesting that there is no obvious structural change in the functionalized NTH.The stability of the functionalized NTH was further examined by considering the structural relaxation under isothermal-isobaric ensemble at 300 K.As compared in Fig.1d,the potential energy of the sample fluctuates around a certain value,signifying the stability of the structure.
The tensile testing was carried on the sample with a length of about 24 nm.The model was firstly optimized by the conjugate gradient minimization method and then equilibrated under the isothermalisobaric ensemble using Nos′e-Hoover thermostat and barostat [40,41]for 500 ps.During the structural relaxation,periodic boundary conditions were applied along the length direction(i.e.,connected end to end).The radial distribution function was calculated under the microcanonical ensemble for another 1 ns simulation.After structural relaxation,the model was switched to non-periodic boundary conditions.One unit cell at the end of the NTH was fixed,and a constant velocity of 0.005 Å/ps was applied to one unit cell at the other end of the NTH to perform tensile deformation.These two unit cells were treated as rigid body during the tensile deformation,and a linear velocity gradient was applied to the whole deformation region to ensure uniform deformation.A small time step of 0.5 fs was used in the simulation.For the given tensile load,the NTH will be stretched by 2.5×10-6Å by a single simulation step(or 0.5 fs).To reduce the influence from thermal fluctuations,a low temperature of 10 K was employed in all simulations,which was commonly adopted in literature.All simulations were performed by the open-source package LAMMPS[42].
During the simulation,the virial stress was calculated according to Ref.[43].

Fig.2.Stress-strain curves of NTH with different percentages of ethyl functional groups.
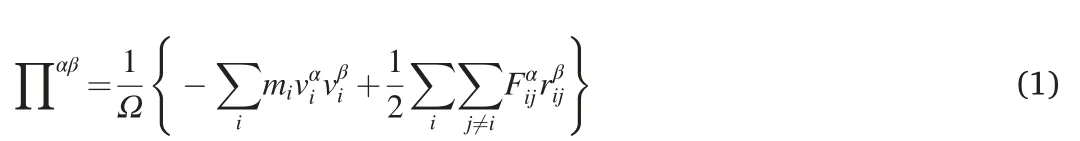
Here,Ω represents the volume of the model;miandviare the mass and velocity of atomi,respectively;Fijandrijare the force and distance between atomsiandj;and the indices α and β represent the Cartesian components.Following previous works [19,21],the (3,0) NTH was approximated as an ultra-thin cylinder with a diameter of 0.5 nm.With this approximation,the atomic virial stress can be estimated using the effective volume for individual atom.To note that adopting different volume approximation approaches will affect the magnitude of stress,however,it will not influence the discussions and conclusions in this work as they rely on the relative magnitude of the stress.The engineering strain as calculated from ε =Δl/l0was used,where Δlandl0represent the length change and the initial length of the NTH,respectively.
Additional first principles calculations were carried out to assess the bond configuration of the NTH with functional groups based on VASP[44].The generalized gradient approximation (GGA) proposed by Perdew-Burke-Ernzerhof (PBE) exchange-correlation potential [45] was adopted.Basically,three unit cells of the (3,0) NTH was adopted as the super cell,and one functional group was introduced in the middle of the super cell.The cut-off energy for plane wave was set to 500 eV.The energy criterion was set to 10-5eV in iterative solution of the Kohn-Sham equation.The Brillouin zone integration was performed using a 1×1×5 k-mesh.
3.Results and discussions
3.1.Tensile deformation of NTH with ethyl functional group
We first assess the tensile behaviours of the NTH with randomly dispersed ethyl groups.As compared in Fig.2,the ethyl functionalized NTH share a similar brittle feature as the pristine NTH,i.e.,the stress increases monotonically until a sudden drop to zero,corresponding to a brittle failure (when bond breakage occurs).Unexpectedly,the presentences of ethyl groups degrade the tensile properties in a large extent,in terms of fracture strain/strength.Here,the fracture strain refers to the strain threshold when bond breakage occurs,and the corresponding stress is taken as the fracture strength.In detail,the pristine NTH has a fracture strain of about 13.50%with a fracture strength of~87.22 GPa.However,the NTH with 6% of ethyl functional groups shows a fracture strain of about 8.82%,which is around 35% reduction.Compared with the fracture strain/strength,Young's modulus experiences a much smaller degradation.Here,Young's modulus is estimated from the linear fitting for elastic deformation regime with a strain within 1.5%.For the pristine NTH,Young's modulus is about 1092 GPa,which agrees well with the literature[20,21].The NTH with 6%of ethyl functional groups exhibits a Young's modulus of about 1032 GPa,corresponding to a reduction of around 5%.
The remarkably suppressed fracture strain can be explained from the atomic configurations.As illustrated in Fig.3a,the atomic stress is uniformly distributed in the pristine NTH during the whole tensile deformation process.However,there is a clear stress concentration at the C atoms where the ethyl functional group is introduced(Fig.3b).With the increase of strain,bond breakage is observed at the stress concentration location,which leads to the eventual failure of the NTH.The stress concentration also explains the observation that even a small content of functional groups (1%,i.e.,6 functional groups) can obviously degrade the tensile properties of NTH as shown in Fig.2.
3.2.Influence from different types of functional groups
In literature,different functional groups have been used to modify CNTs,such as–(CH2)n–,EPON-862 (epoxy resin),–(C6H4)n–,and aromatic links [46–48].Thus,it is interesting to know how different functional groups might influence the tensile properties of NTH.For such purpose,we adopt another two types of functional types,including phenyl group (-C6H5) and methyl group (-CH3).Specifically,the tensile behaviors of NTH with 6%of functional groups are examined.For a fair comparison,the locations of functional groups are kept the same while adding different types of functional groups,which is controlled by the same random seed number using our house-code.As illustrated in Fig.4a,the functionalized NTH share a similar radial distribution function,indicating insignificant structural changes after adding different types of functional groups.For the NTH with phenyl functional group(the dotted lines in Fig.4a),a new peak (~1.35 Å) is observed between the C–H bonds and the sp3C–C bonds,which correlates to the sp2bonds in the phenyl functional group.
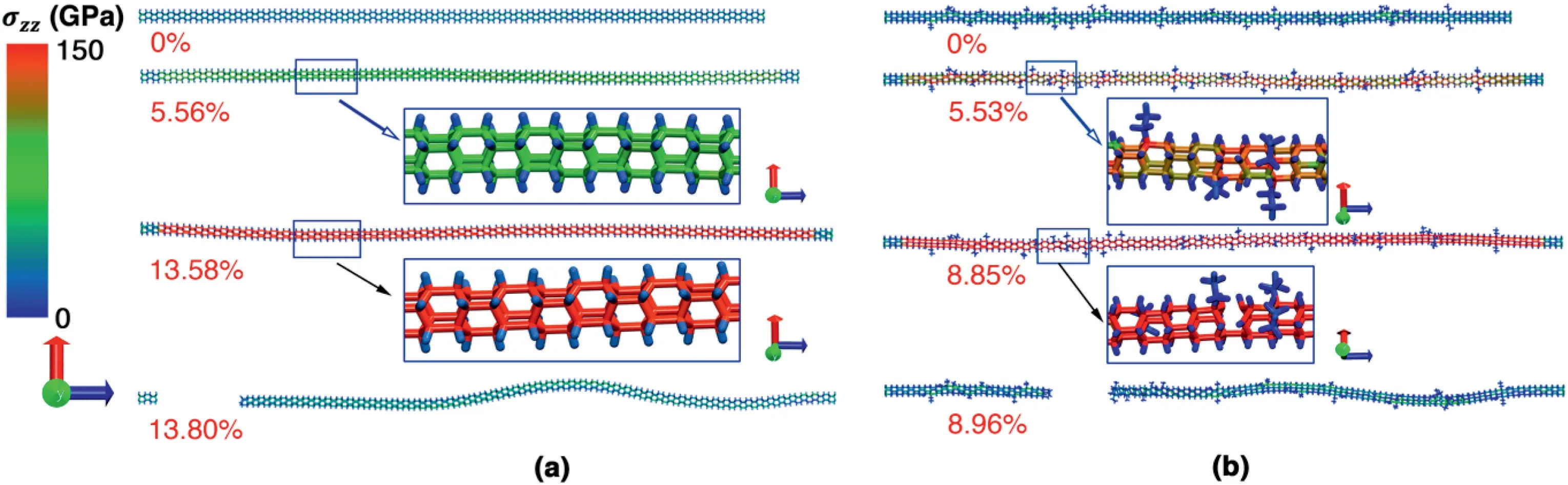
Fig.3.Atomic configurations at different tensile strains.(a) The pristine NTH;and (b) NTH with 6% of ethyl groups.
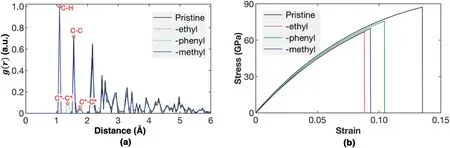
Fig.4.Tensile deformation of NTH with 6%of functional groups.(a)The radial distribution function of the NTH with different functional groups;and(b)The stressstrain curves for the NTH with different functional groups.
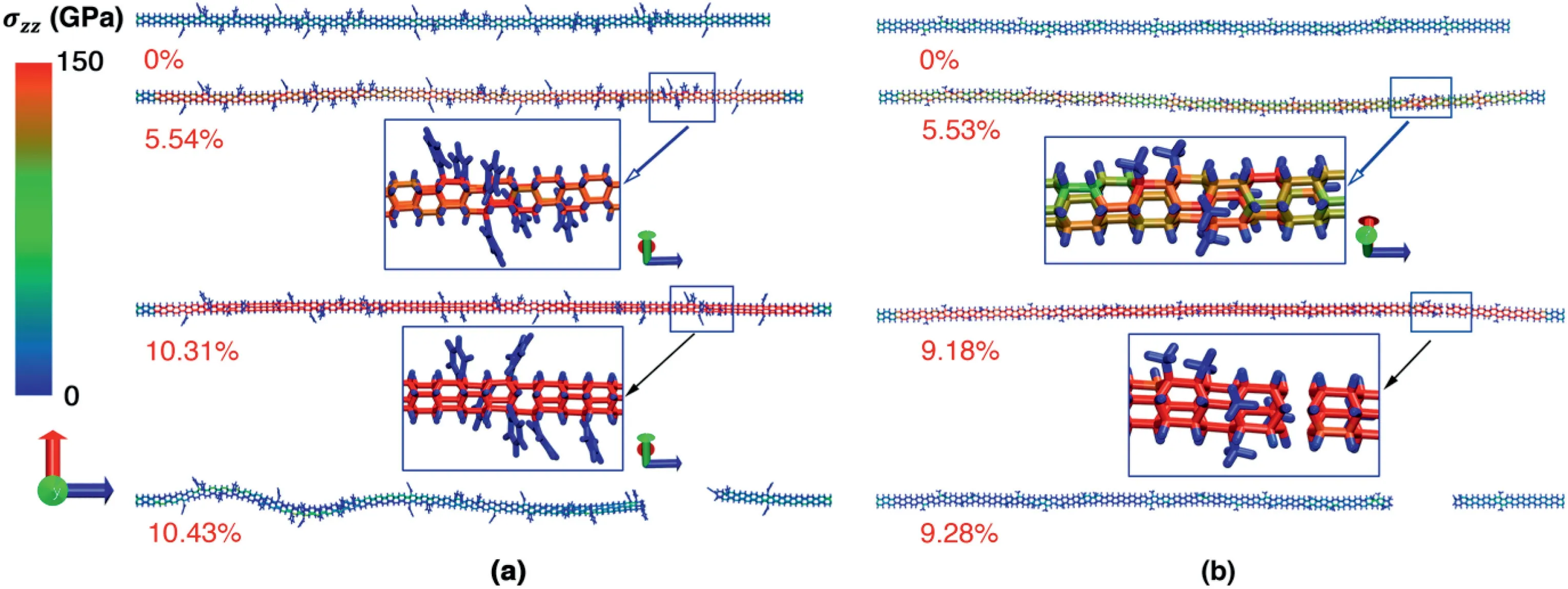
Fig.5.Atomic configurations at different tensile strains.(a) The NTH with 6% phenyl groups;and (b) NTH with 6% of methyl groups.
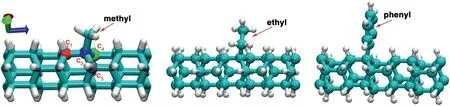
Fig.6.Bond configurations of the NTH super cell with one functional group after structural optimization from first principal calculations.
As compared in Fig.4b,the impacts on the tensile properties of NTH from different functional groups vary largely from each other.The fracture strain is about 10.41% and 9.28% for the NTH with phenyl and methyl functional groups,respectively,corresponding to a reduction of about 23% and 31% compared with the pristine NTH,respectively.Similar degradation applies to the fracture strength.These results suggest that ethyl and methyl groups result in stronger influence on the tensile properties of NTH than that induced by phenyl groups.Similar with the ethyl functionalized NTH,the phenyl and methyl groups result in insignificant impacts on Young's modulus,which is about 1060 GPa and 1049 GPa,respectively.
To exploit the mechanisms underlying the impacts from different functional groups,we compared the atomic configurations at different strain magnitudes.As shown in Fig.5a,the NTH also exhibits a clear stress concentration at the C atoms where phenyl groups are introduced,which becomes the potential bond breakage site.The eventual failure is found at the section where severe stress concentration is observed due to the presence of several phenyl groups.Similar deformation process is also detected for the NTH with methyl groups(Fig.5b).From Figs.3 and 5,it is uniformly found that bond breakage occurs at the C–C bonds that are adjacent to the functional site.
Above results reveal that the addition of functional groups uniformly results in local stress concentration,which degrades the tensile properties of the NTH.To further assess the impacts from different functional groups,we compare the resulting bond changes in Fig.6 as acquired by first principles calculations.After energy minimization,the presence of functional group is found to alter the local bond configuration slightly.For discussion convenience,the four C atoms adjacent to the functional site are denoted asc1toc4,respectively,andcicjand ∠cicjckrepresent the corresponding bond and angle,respectively.For the pristine structure,c1c2is about 1.58 Å.c2c3is the same asc2c4,which is about 1.546 Å.The angle is the same for ∠c1c2c3and ∠c1c2c4,which is about 112.4°.The ∠c3c2c4angle is slightly smaller,about 106.4°.As summarized in Table 1,different functional groups lead to different bond changes.It is uniformly found that the adjacent C–C bonds are stretched due to the presence of functional group.The phenyl group results in the largest bond length change of about 1.39%forc1c2bond,while the methyl group induces the largest bond length change of about 1.0% forc2c3orc2c4bond.These bond length changes are accompanied by angle changes.The initial symmetry betweenc3andc4is generally maintained when phenyl or methyl group is introduced,as indicated by the similar ∠c1c2c3and ∠c1c2c4angles.However,for ethyl group,these two angles exhibit a difference of about 2.7°,signifying a relatively large local bond distortion.Such large bond distortion is regarded as one of the explanations for a stronger degradation effect from ethyl group shown in Fig.4b.It is worthy to note that the interfacial C bonds at the functional site for–CH3and–C2H5groups are sp3-sp3bonds (about 1.54 Å),which is slightly longer than that for–C6H5groups in theory (i.e.,sp3-sp2bonds with a length of about 1.50 Å).A longer interfacial C bond may also contribute to the stronger degradation effect from methyl or ethyl groups.
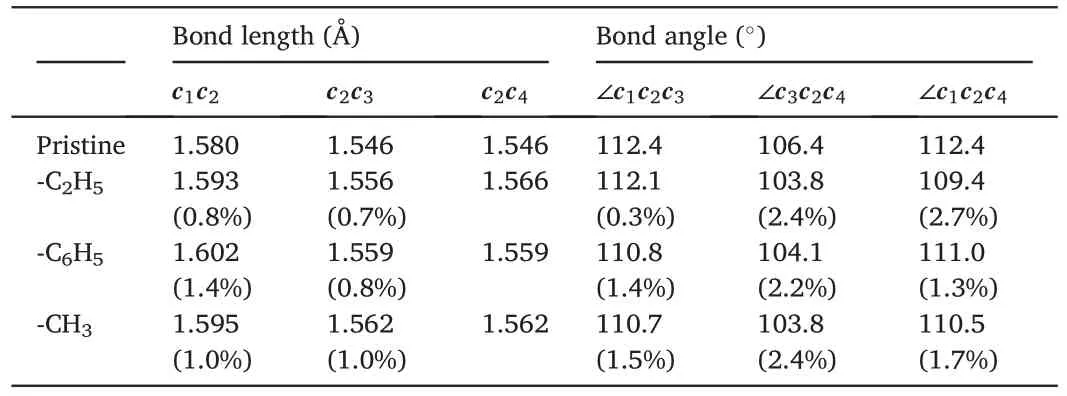
Table 1 The carbon bond and angle at the functional site from first principles calculation.The values in brackets are the relative difference compared with that of the pristine NTH.
3.3.Influence from the content and distribution of functional groups
With above understanding,we then investigate how the content of functional groups will affect the NTH's tensile properties.For such purpose,we consider a functionalization percentage up to 14%,which is selected to avoid two functional groups in one C–C bonds.In addition,the model with a higher percentage of functional groups contains all functional sites in a lower percentage.Fig.7a compares the tensile properties of NTH with different percentage of ethyl groups.In general,the NTH with a higher percentage of functional groups exhibits a lower fracture strain/strength and a smaller Young's modulus.It is found that Young's modulus decreases monotonically when the content of functional groups increases.However,the fracture strain does not exhibit a uniform decreasing trend.For example,the NTH with 8%of ethyl groups shows a higher fracture strain than its counterpart with 4%of ethyl groups.Such observation is expected as originated from a synergetic effect from sample length and stress concentration,similar as seen in the NTH containing Stone-Wales transformation defects[21].

Fig.7.Tensile properties of NTH with different contents of ethyl groups.(a)The fracture strain and Young's modulus as a function of the functional percentage;(b)The self-oscillation of the NTH during tensile deformation,including the positions of the center of mass (left panel),and the trajectory of the center of mass as a function of time;(c)End view of the C atom distribution for the functionalized NTH;and(d)Tensile properties of the NTH with a same percentage of ethyl groups but different distribution patterns.
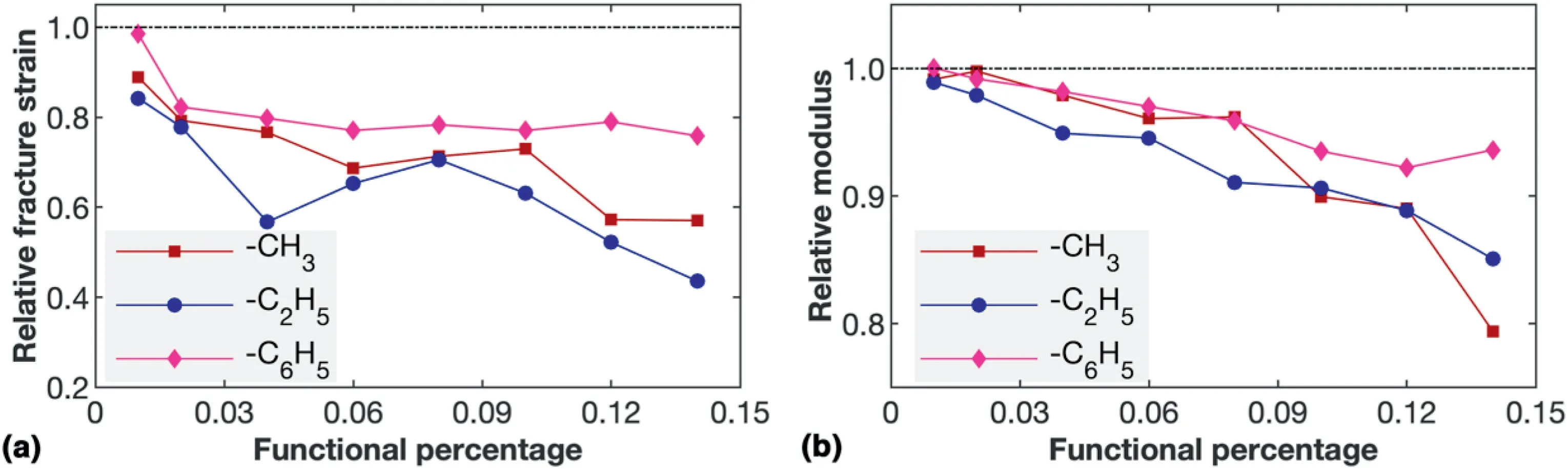
Fig.8.Tensile properties of functionalized NTH.(a)The relative fracture strain in related to the pristine NTH as a function of the percentage of functional groups;and(b) The relative Young's modulus as a function of the percentage of functional groups.
Theoretically,the fracture can occur at any stress concentration sites during the tensile deformation.The models with higher percentage of functional groups possess different overall bond distortion scenarios,which will affect the stress concentration locations.Meanwhile,due to the dynamic nature of the atomic system,NTH will oscillate in the lateral directions during the tensile process.As demonstrated in Fig.7b,the functionalized NTH exhibits a much larger oscillation magnitudecompared with that of the pristine NTH during tensile deformation.Here,the oscillation magnitude is calculated from,withandrepresenting center of mass of the NTH.The oscillation has a strong dependence on the sample length and the distribution pattern of the functional groups,which will alter the instantaneous stress distribution during deformation.It is found that all examined ethyl functionalized NTHs show a much larger oscillation magnitude than their pristine counterpart,except the NTHs with 6%of ethyl groups(which is similar as that of the pristine NTH).An uneven mass distribution is expected to promote the self-oscillation during deformation.As affirmed from the C atom distribution analysis in Fig.7c,the NTH with 6%of ethyl groups has a much even C atom distribution in the radial direction compared with the sample with 4%of ethyl groups.Overall,with the synergetic impacts from the stress concentration and self-oscillation during tensile deformation,some models with a lower functional group content may exhibit a smaller fracture strain compared with the sample with more functional groups.
To further illustrate the influence from the locations of stress concentration,we compare five NTH models containing 8% ethyl groups with different random distribution patterns,denoted as model I to model V.Refer to the first principles calculation results in Fig.6,the addition of functional groups will alter the local bond configuration.Therefore,the stress concentration sites will vary with the distribution pattern of the functional groups.As compared in Fig.7d,a large variation is observed for the tensile properties between these five models,though they possess a same content of ethyl groups(same total atom number).It is worthy to mention that model II show excellent tensile properties comparable with that of the pristine NTH with ignorable degradation on fracture strain/strength (~0.4%).It is suspected that a proper distribution of the functional groups could alleviate the stress concentration and thus maintain the excellent mechanical properties of NTH,though a future investigation is required for more confined models.Overall,these results suggest that the influence from the functional groups is not only related with their content,but also relies on their distribution pattern.
Before concluding,we also compute the tensile properties of NTH with different percentages of phenyl and methyl groups.For a fair comparison,the functional sites are kept the same for a same functional percentage.As plotted in Fig.8a,ethyl groups are found to introduce stronger degradation effect to the tensile properties of NTH in the examined functionalization ratio,followed by methyl groups.A huge degradation of around 60% for the fracture strain is observed for the samples with 14% of ethyl groups.In comparison,the degradation on Young's modulus is not that significant(Fig.8b).These obversions align with the fact that adding functional groups will not change the sp3bond nature of the NTH,however,they will induce local stress concentrations.As a result,Young's modulus experiences a much smaller influence from the functional groups as compared with the fracture strain or fracture strength.
4.Conclusions
Based on large scale molecular dynamics simulations,this work exploits the impacts on the tensile properties of NTH from randomly dispersed functional groups.It is found the introduction of functional groups will alter the local bond configuration,i.e.,stretching the adjacent sp3C bonds and change the C bond angles of the NTH,which results in initial stress concentration.Such stress concentration becomes the bond breakage sites during tensile deformation,which significantly reduces the fracture strain/strength of the NTH.For illustration,1% of ethyl functional group is found to introduce~16%of reduction on the fracture strain.Despite that,functional groups exert a smaller impact on Young's modulus.Simulation results reveal that the impacts on the tensile properties of NTH varies when functional type changes.Such observation can be explained by the atomic configurations,from where different functional types are found to results in different local bond changes.Further simulations show that Young's modulus decreases monotonically when the content of functional groups increases,which however is not the case for the fracture strain/strength.It is found that some NTHs with higher content of functional groups exhibits higher fracture strain/strength than their counterparts with lower content of functional groups.Such surprising observations are anticipated as attributed to the synergetic effects from the length effect,distribution pattern of functional groups and selfoscillation of NTH during the tensile deformation.That is,the NTH will undergo self-oscillation,which will alter the instantaneous stress distribution and influence the occurrence of bond breakage or fracture.It is further shown that the NTHs with a same content of functional groups can exhibit different tensile behaviours when the distribution pattern is different,and the NTH with even distributed functional groups can maintain its excellent tensile properties.
In general,it is found that ethyl groups induce stronger degradation on the tensile properties of NTH,followed by methyl and phenyl groups.The influence from the functional groups can be further exploited by examining the phonon instability based on first principles calculations,which deserves a separate investigation.Literatures already show that phonon instability occur for low dimensional nanostructures before fracture[49],such as graphene[50],silicene[51],and borophene[52].Overall,this work provides a comprehensive understanding of the influence from functional groups on the mechanical properties of NTH,which should be beneficial to their engineering applications.
Author contributions
HZ designed the models and carried out the simulations.JS carried out the first principles calculations.All authors contribute to the analysis,discussion and writing.
Declaration of competing interest
The authors declare that they have no conflict of interest.
Data availability
The data that support the findings of this study are available from the corresponding authors on reasonable request.
Acknowledgement
Support from the ARC Discovery Project (DP180103009,DP200102546) and the High-Performance Computing (HPC) resources provided by the Queensland University of Technology(QUT) are gratefully acknowledged.

- Namo Materials Science的其它文章
- Fabrication of segregated poly(arylene sulfide sulfone)/graphene nanoplate composites reinforced by polymer fibers for electromagnetic interference shielding
- In situ polymerization preparation and mechanical properties of nanocomposites based on PA10T/10I-block-PEG copolymer and graphene oxide
- CNT toughened aluminium and CFRP interface for strong adhesive bonding
- Abnormal enhancement to the quality factors of carbon nanotube via defects engineering
- Fracture behavior of hybrid epoxy nanocomposites based on multi-walled carbon nanotube and core-shell rubber
- Anti-corrosion and electrically conductive inorganic conversion coatings based on aligned graphene derivatives by electrodeposition