
SPRED1基因突变致Legius综合征1例病例报告
2022-10-07 07:01:44任韵清丘贵英刘吉鹏吴鼎文
中国循证儿科杂志 2022年4期
任韵清 丘贵英 刘吉鹏 吴鼎文
1 病例资料
女,2岁。因“躯干、四肢多发褐色斑片2年”就诊于浙江大学医学院附属儿童医院(我院)皮肤科门诊。患儿出生时躯干、四肢即有多发褐色斑片,随年龄增长数量逐渐增多、面积增大,无疼痛、瘙痒等不适。患儿足月自然分娩,无出生窒息抢救史,既往体检未见异常,智力、运动发育正常,无惊厥抽搐史,无过敏史,无重大疾病史。父母体健,非近亲婚配,否认遗传代谢性疾病史。患儿父亲躯干、四肢见散在褐色斑片,性质与患儿一致,余未见异常。
查体:一般情况可,营养状况可,未见特殊面容,头围47.3 cm,躯干、肢体发育正常,心、肺、腹检查未见异常。颈部、躯干和四肢呈多发性圆形、椭圆形或不规则形褐色斑片,直径0.2~6 cm,散在分布,未见皮肤赘生物及虹膜Lisch结节(图1)。
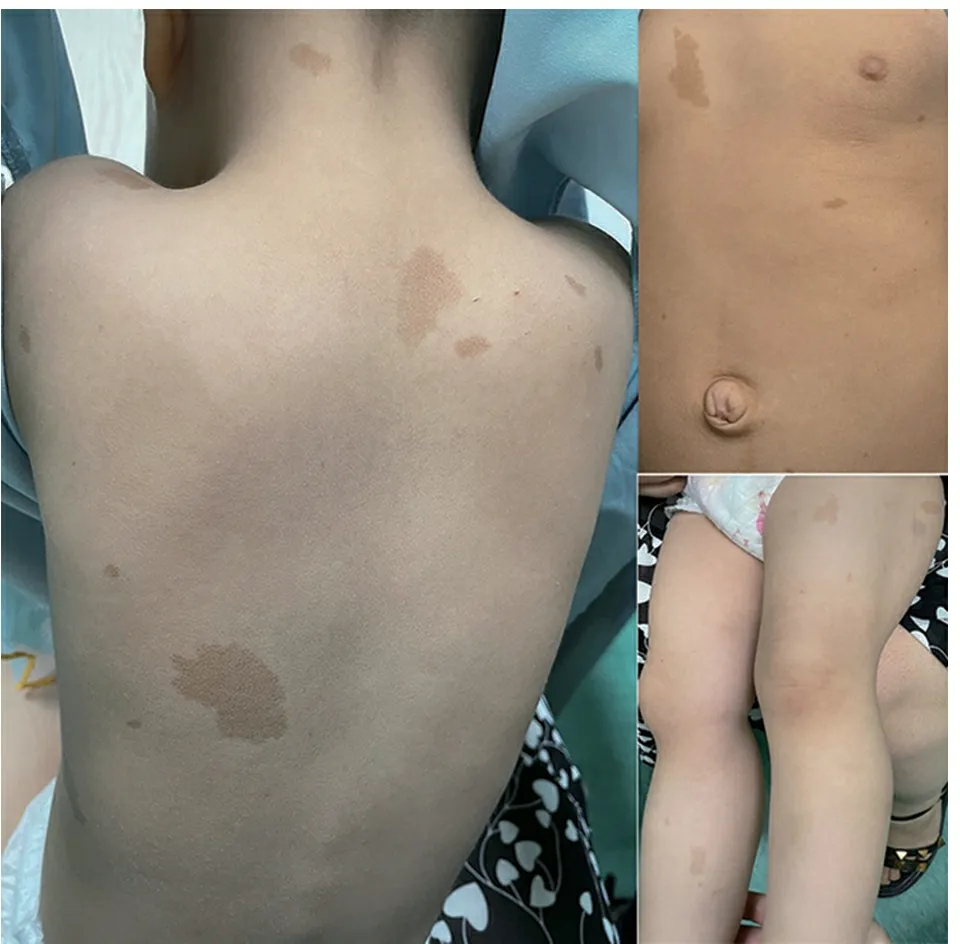
图1 患儿躯干和四肢不规则或椭圆形咖啡牛奶斑
实验室检查:血、尿常规、心脏多普勒超声、头颅MR检查未见异常。
基因检测:经我院伦理委员会批准(审批号:2022-IRB-046),取得患儿父母的知情同意后,采集患儿外周血2 mL送基因检测,患儿父母拒绝行基因检测。使用QIAamp DNA Blood Mini Kit提取基因组DNA。采用安捷伦公司ClearSeq Inherited Disease外显子序列捕获试剂盒(含2 742个基因),通过多重探针杂交方法靶向富集目标区域序列,在HiSeq 2500(Illumina)平台进行二代测序。目标区域包括目标基因的外显子及其邻近±10 bp内含子区域。测序获得的DNA序列与人类基因组hg19参考序列进行比对,对覆盖度≥20×的变异进行生物信息学分析和致病性分析。数据解读规则参考美国医学遗传学和基因组学学院相关指南。结果显示患儿存在SPRED1(NM_152594.3)基因exon 1的起始密码子变异c.3G>A(p.M1I)。SIFT、Polyphen及MutationTaster等软件行蛋白功能预测,提示为“中度危害”。此突变在ClinVar数据库对其注释为“可能致病的”,在HGMD注释为“致病的”。根据先证者的临床表型和基因检测结果,确诊为Legius综合征(LGSS),且错义突变c.3G>A具有致病性。
2 讨论
LGSS又称Ⅰ型神经纤维瘤样综合征,是由SPRED1基因突变引起的常染色体显性遗传病,患病率为1/75 000~1/46 000[1],迄今为止国内外仅报道百余例(中国2例)[2-4]。SPRED1基因位于15q13,长约104.4 kb,由7个外显子构成。其编码的SPRED1蛋白由444个氨基酸构成,包含N端EVH-1结构域、中央c-Kit结合结构域和C-末端SROUTY结构域[5]。SPRED1蛋白通过负调控Ras-MAPK通路抑制生长因子、激素、细胞间的相互作用,进而抑制细胞的增殖、存活、分化、迁移或代谢。当SPRED1基因发生突变时,其功能丧失或产生缺陷,导致颅面部畸形、皮肤、神经和心血管受累等[6]。
SPRED1基因突变最早由Pasmant等[7]报道,已发现的突变类型主要包括无义突变、移码突变、错义突变和大片段缺失突变等[2],目前尚无突变热点的相关分析。本文病例检测到SPRED1基因起始密码子突变c.3G>A(p.M1I),该突变在Clinvar和HGMD数据库中分别评估为“可能致病的”和“有害的”;患儿父亲也有类似的临床表型,但因其拒绝行基因检测,故无法得知其家系情况。
LGSS临床表现与Ⅰ型神经纤维瘤(NF1)相似但较轻,以皮肤多发咖啡牛奶斑、伴或不伴皱褶部位(腋窝和腹股沟)的雀斑为主要特征,其余表型还包括身材矮小、巨头畸形、多发性脂肪瘤、学习障碍、语言延迟、注意力缺陷、多动障碍、发育迟缓等。由于LGSS与NF1的临床症状重合率极高,临床上鉴别较为困难。Pasmant等[1]对 279例表现为多发性咖啡牛奶斑的患者进行了二代测序分析,其中LGSS占4%。与NF1相比,LGSS患儿中学习困难、注意力缺陷的发生率及肿瘤易感性较低,无NF1相关肿瘤和特征性骨病变,如神经纤维瘤、视神经胶质瘤、Lisch结节、蝶骨翼发育不良和假关节形成等。研究发现,NF1的基因产物神经纤维蛋白通过与SPRED1蛋白的EVH-1结构域结合而抑制下游的RAS-MAPK通路,调节细胞的生长和增殖,这可能是NF1和LGSS临床表型高度重合的关键机制[8]。嵌合型LGSS由患者局部组织携带SPRED1基因突变引起,其与典型LGSS不同的是,表现为躯体节段性病变,且突变仅能在病变组织中测出[9]。Legius等[10]根据临床症状和实验室检查结果对典型LGSS和嵌合型LGSS的诊断标准进行了修订(表1)。

表1 典型Legius综合征(LGSS)和嵌合型LGSS的诊断标准[9]
LGSS的基因型-表型关系目前尚不明确。①LGSS可能有累及血管的风险。Pabst等[11]报告了1例合并Moyamoya综合征的LGSS患者,表现为颈内动脉狭窄引起的脑缺血症状,如左侧肢体无力、左侧面部感觉减退和持续性头痛等;基因检测发现SPRED1基因1号外显子和1号内含子之间40.51 kb的小片段缺失突变。Romanisio等[12]也描述了1例合并Moyamoya综合征的20岁LGSS女性患者,其携带SPRED1基因c.330_331delGG缺失突变;临床表现为左侧偏瘫、运动无力和全面性强直阵挛发作,与文献[11]类似。因此,应在LGSS患者中行相关评估(如尽早完善MRA)。②目前发现LGSS患者并发的肿瘤包括横纹肌肉瘤、神经母细胞瘤、淋巴母细胞白血病、急性髓母细胞白血病和膀胱癌,但无法确认这些肿瘤的发生与LGSS相关[13]。③通常认为LGSS无虹膜Lisch结节表现,但近年文献报道了2例LGSS姐妹也出现了双侧虹膜Lisch结节的现象[14]。
LGSS目前尚无有效的治疗方法,皮肤症状如咖啡牛奶斑和雀斑可采取激光治疗。Borrie等[15]在SPRED1基因双敲除的小鼠模型中观察到了认知障碍和孤独症谱系障碍,通过MEK抑制剂PD325901对Ras-MAPK通路进行靶向治疗,小鼠的相应症状有明显好转甚至完全消退。MEK抑制剂目前在临床上用于治疗NF1患者的症状性和无法手术的丛状神经纤维瘤。在NF1的青少年和成人患者中的2期试验显示,PD325901可使神经纤维瘤体积缩小,有望用于治疗LGSS。本例患儿仅表现为全身多发性咖啡斑,因美观需求,颈部3处咖啡牛奶斑行调Q694 nm红宝石激光治疗1次,自觉消退不明显,故未再次治疗。目前随访半年,咖啡斑未见明显增多,尚未发现多发性脂肪瘤、语言延迟、注意力缺陷、多动障碍、发育迟缓等表现。
猜你喜欢
中华养生保健(2020年7期)2020-11-16 01:14:28
诗选刊(2019年9期)2019-11-20 10:24:01
现代园艺(2017年21期)2018-01-03 06:41:32
高中生·天天向上(2017年4期)2017-06-09 02:37:17
中国康复理论与实践(2015年10期)2015-12-24 05:42:44
中国医疗美容(2015年1期)2015-07-12 10:06:04
医学研究杂志(2015年8期)2015-06-22 14:00:56
医学研究杂志(2015年5期)2015-06-10 06:43:26
中国康复理论与实践(2015年7期)2015-05-09 08:31:46
现代检验医学杂志(2015年5期)2015-02-06 01:42:20
