
CexZr1−xOy 常温催化氧化去除甲醛的研究
2022-09-21 12:03何人广谭小芳贾丽娟段开娇高冀云刘天成
云南大学学报(自然科学版) 2022年5期
刘 巷,何人广,谭小芳,贾丽娟,段开娇,高冀云,常 玉,刘天成**
(1.云南民族大学 化学与环境学院;云南省高校工业废气绿色净化技术科技创新团队,云南 昆明 650504;2.云南技师学院,云南 昆明 650500)
甲醛(挥发性有机物)与NOx、SO2、颗粒物等相互作用后导致光化学烟雾等环境问题的发生[1].人们长期接触低剂量甲醛会引起慢性中毒、鼻咽癌、白血病等[2].甲醛的强黏合性使其被用于胶合剂的生产[3],而居室装修中又利用胶合剂将人造板粘合起来[4].在一定温度湿度条件下,板材会释放出一定量的甲醛[5].新装修室内环境下,1 h 甲醛质量浓度可高达0.121 mg/m3[6],超过室内空气质量标准0.08 mg/m3[7].目前室内甲醛的无害化处理技术有物理法[8]、生物法[9]、物理化学吸附法[10-11]等,但这些方法效率低、过程复杂.催化剂催化氧化甲醛是较为高效、简便的方法.例如,卢素红等[12]制备Co3O4/Al2O3催化剂,在80 ℃下将甲醛完全转化.Fang 等[13]制备了MnOx/AC 甲醇催化剂,研究表明,在1 000 min 范围内,甲醛的去除率基本保持在100%.
铈锆固溶体是一种优异的材料[14-15],广泛应用于催化氧化领域.CeO2在高温下易烧结,热稳定性较差.通过在CeO2中掺入ZrO2,可增强催化剂的热稳定性,显著提升其催化性能[16].例如,孙宇等[17]利用铈锆固溶体选择性催化氧化H2S,硫收率达到97%.黄嘉苗等[18]制备Cu/Ce0.75Zr0.25O2催化剂催化乙醇干气重整反应,乙醇转化率为100%.
本文利用共沉淀法制备CexZr1−xOy催化剂,考察其在常温下对甲醛的催化性能,结合表征结果分析铈锆摩尔比对甲醛去除率的影响,探究CexZr1−xOy失活的原因,为常温除甲醛提供一定的参考.
1 实验部分
1.1 实验仪器及试剂
1.1.1 仪器 SHZ-D(Ⅲ)循环水式真空泵,SZCL−2 数显智能控温磁力搅拌器,巩义市予华仪器有限责任公司;D08−4F 质量流量显示仪,D07−19B 质量流量计,北京七星华创科技有限公司;ϕ4.5 cm×100 cm 自制石英管.
1.1.2 气体及试剂 N2(φ≥99.999%),O2(φ≥99.999%),云南铜业股份有限公司;甲醛(CH2O),大连大特气体有限公司;Ce(NO3)3·6H2O(A.R.),Zr(NO3)4·5H2O(A.R.),上海麦克林生化科技有限公司;水合联氨(A.R.),天津市风船化学试剂有限公司;无水乙醇(A.R.),天津市致远化学试剂有限公司;石英砂(A.R.),天津市科密欧化学试剂有限公司.
1.2 CexZr1-xOy 制备通过共沉淀法分别以n(Ce)/n(Zr)=0,0.2,0.4,0.6,0.8,1.0 制备了6 种CexZr1-xOy催化剂,分别记作ZrO2,Ce0.8Zr0.2Oy,Ce0.6Zr0.4Oy,Ce0.4Zr0.6Oy,Ce0.2Zr0.6Oy,CeO2.具体操作如下:分别称取适量的Ce(NO3)3·6H2O、Zr(NO3)4·5H2O,溶于一定量的去离子水中并搅拌10 min,加入水合肼调节pH 值至10.5.将制备的混合沉淀物搅拌0.5 h,陈化4 h,过滤,所得产物用去离子水及乙醇洗涤,干燥,焙烧,研磨,制得CexZr1-xOy催化剂.
1.3 催化剂表征样品的物相组成利用德国Bruker D8 Advance 型X 射线衍射仪进行测试,测试条件:CuKα,2θ=10º~80º,扫描速率10 º/min,步长0.01º/s.催化剂形貌分析采用美国FEI 公司的Nova Nano SEM450 型场发射扫描电镜,加速电压30 kV,用乙醇将样品分散,烘干后,喷金处理.采用麦克2460型分析仪对催化剂的比表面积、孔径等孔结构参数进行测试,测试前样品在200 ℃下真空处理3 h,在77 K(液氮)条件下进行静态氮吸附,BET 法计算比表面积,BJH 法计算孔容.X 射线光电子能谱表征采用Thermo Scientific K-Alpha 型仪器,AlKα,工作电压为12 kV,结合能使用内标碳1s 峰(Eb=284.80 eV)进行校准,精度±0.2 eV.
1.4 催化剂的活性测试如图1 所示,由实验室搭建的甲醛催化氧化活性评价系统测定样品CexZr1-xOy的催化活性.将0.2 g 催化剂(40~60 目,0.301~0.441 mm)和50 g 石英砂混合均匀装入内径45 mm 的石英管中,在常温常压下通入反应气.采用钢瓶动态配气,各气体的体积分数为:20%O2,60%甲醛(CH2O),N2为平衡气体.利用酚试剂分光光度法检测CH2O 体积分数.由式(1)计算CH2O转化率:
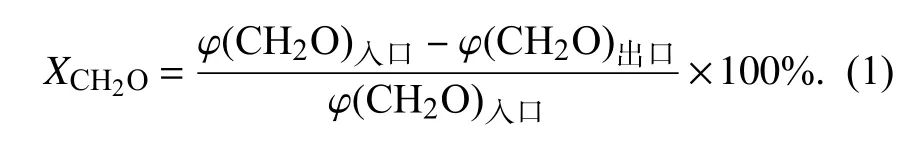
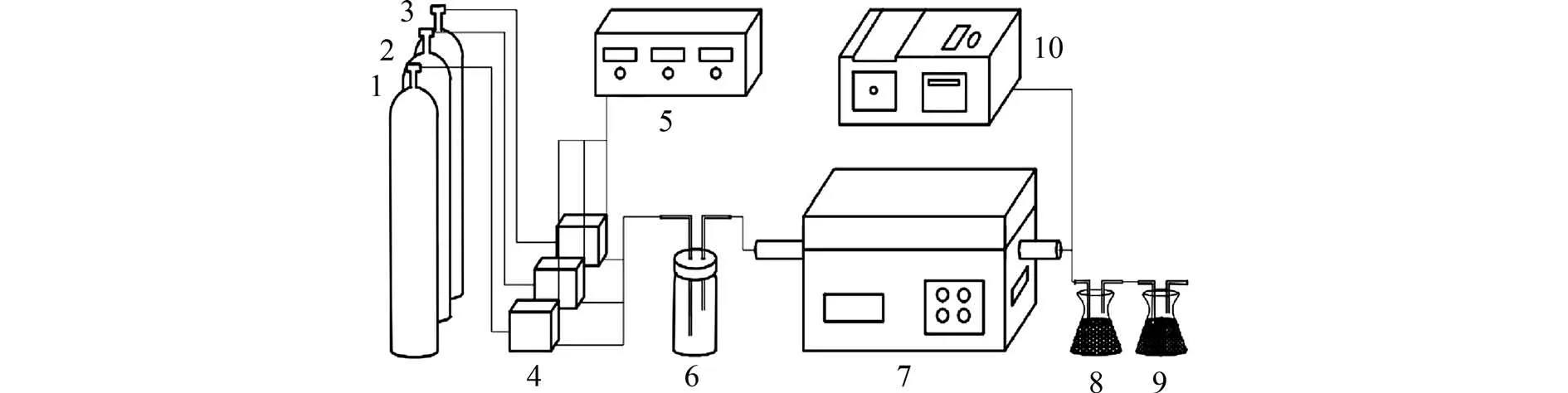
图1 实验流程图Fig.1 Experimental flow chart
2 结果与讨论
2.1 物相分析用XRD 测定催化剂的晶体结构,结果见图2.CeO2表现出立方萤石结构的强烈而尖锐的衍射峰(PDF 43-1002),其(111)、(200)、(220)、(311)、(222)和(400)晶体面分别对应28.5°、33.1°、47.5°、56.3°、59.1°和69.4°处的衍射峰.CexZr1−xOy催化剂表现出更宽的峰,并向更高的2θ转移,表明Zr 被成功地掺杂到二氧化铈晶格结构中[19].计算得到的晶格参数值列于表1.ZrO2的掺入使CeO2发生相位扭曲,半峰宽增加,表明Zr 的掺杂减小了催化剂的晶粒尺寸[20].随着n(Ce)/n(Zr)的增加,2θ值向低角度移动,这是由于Zr4+离子半径(0.084 nm)小于Ce4+的离子半径(0.097 nm)[21],Zr4+掺入晶格后晶面间距缩小[22],从0.312 nm 下降到0.299 nm.这些结果证实了Zr4+掺入CeO2的晶格,形成了铈锆固溶体.图2 的XRD 衍射峰和表1中的晶格参数与Liu 等[23]所揭示的一致.
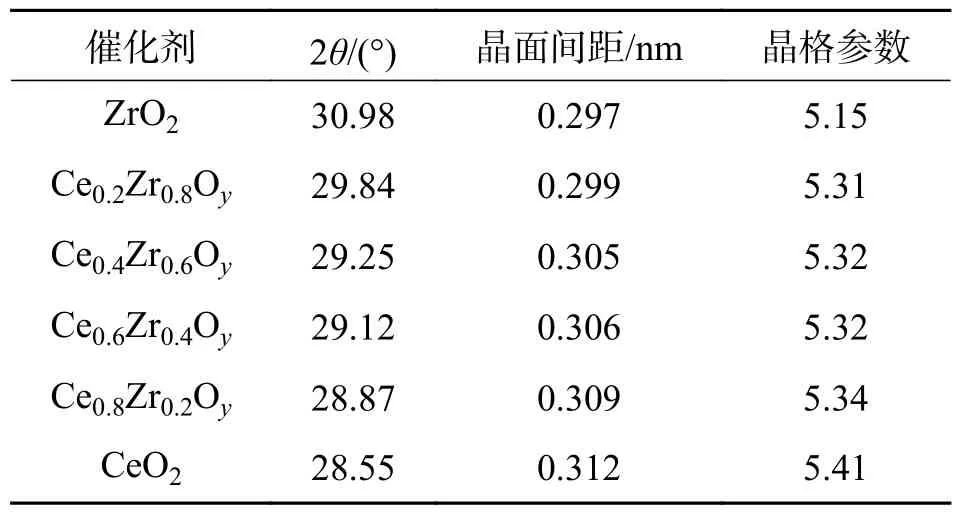
表1 CexZr1−xOy 晶面参数Tab.1 Lattice parameters of CexZr1−xOy

图2 CexZr1−xOy XRD 图Fig.2 XRD diagram of CexZr1−xOy
2.2 形貌分析利用扫描电镜对催化剂的形貌进行观察.如图3 所示,ZrO2为表面光滑的不规则立方体结构,轮廓较为清晰,仅个别晶粒表面有少许裂纹.而CeO2表面粗糙,结构不规则.随着Ce 的负载,CexZr1-xOy催化剂依旧为不规则立方体结构,但Ce0.2Zr0.4Oy、Ce0.4Zr0.6Oy两种催化剂较光滑的表面上出现了明显的裂纹,而Ce0.6Zr0.4Oy催化剂的表面略为粗糙,裂纹也更为明显.Ce0.8Zr0.2Oy催化剂虽与CeO2都呈现蜂窝状的形貌特征,但Ce0.8Zr0.2Oy表面孔的数量远大于CeO2,这种疏松多孔的形貌更容易吸附CH2O,使其被氧化.

图3 CexZr1−xOy 扫描电镜图Fig.3 SEM image of CexZr1−xOy
2.3 结构分析为了探讨n(Ce)/n(Zr)对催化剂比表面积及孔径分布的影响,利用N2吸脱附等温线对CexZr1−xOy进行了测定.图4 为CexZr1−xOy催化剂的N2吸附−脱附曲线和BJH 方法计算的孔径分布曲线,表2 为CexZr1−xOy催化剂的孔结构参数.由图4 可知,CexZr1-xOy均为典型的Ⅳ型等温线,当x>0.5 时,具有H1 型回滞环;随着Zr 的掺入,回滞环逐渐变为H2 型[24].由表2 可知,CexZr1−xOy的比表面积(48.8~59.6 m2/g)、孔体积(0.046 9~0.112 cm3/g)远大于ZrO2(3.10 m2/g、0.006 03 cm3/g),这表明Ce 的负载及Ce 的添加量对于样品的比表面积及孔径尺寸的影响较大.CexZr1−xOy比表面积大于CeO2,推测是Zr4+的掺入增强了催化剂的热稳定性,使其更容易保持较高的比表面积[25].CexZr1-xOy催化剂平均孔径为3.53~10.7 nm,随着n(Ce)/n(Zr)的增加,孔体积与平均孔径整体上呈现增加的趋势.这可能是由于在铈锆固溶体形成过程中,Ce4+尺寸较大,导致晶格膨胀和孔结构的改变.较大的孔径与孔体积可能有利于CH2O 在催化剂表面的扩散和传质,从而提高催化活性[26].

表2 CexZr1-xOy 结构参数Tab.2 Structure parameters of CexZr1-xOy

图4 CexZr1−xOyN2 吸脱附等温线和孔径分布图Fig.4 Nitrogen adsorption-desorption isotherms and pore size distribution of CexZr1−xOy
2.4 表面价态分析采用XPS 对催化剂表面的化学态进行分析.如图5 所示,图中U 与V 分别对应Ce 3d3/2和Ce 3d5/2的自旋分裂轨道.由图5 可知,Ce 3d 轨道可以被分峰为8 个峰,出峰位置分别在:916.08 eV(U4),906.58 eV(U3),902.34 eV(U2),900.28 eV(U1),897.82 eV(V4),888.28 eV(V3),883.9 eV(V2)和881.69 eV(V1).图5 中位于883.9 eV(V2)、902.34 eV(U2)处的峰归属于Ce3+,而位于897.82 eV(V4)、888.28 eV(V3)、881.69 eV(V1)、916.08 eV(U4)、906.58 eV(U3)、900.28 eV(U1)处的峰归属于Ce4+[27].通过峰面积进行半定量计算,可以得出Ce3+占Ce3++Ce4+的摩尔分数,结果列于表3中.由表3 可得,Ce0.8Zr0.2Oy、Ce0.6Zr0.4Oy、Ce0.4Zr0.6Oy、Ce0.2Zr0.6Oy中Ce3+的摩尔分数分别为12.28%、11.81%、14.15%、30.16%.O 1s 轨道可分为在528.74 eV(O1)附近、531.5 eV(O2)附近和532.88 eV(O3)附近3 个峰,分别对应晶格氧、氧空位上的化学吸附氧和分子吸附氧[28].根据峰面积进行定量计算得到,Ce0.8Zr0.2Oy、Ce0.6Zr0.4Oy、Ce0.4Zr0.6Oy、Ce0.2Zr0.6Oy化学吸附氧的摩尔分数分别为83.2%、50.58%、41.18%、34.24%,化学吸附氧随着Ce4+的浓度增大而增加.

图5 CexZr1−xOy XPS 谱图Fig.5 XPS spectra of CexZr1−xOy
由图6 和表3 得出,反应后的Ce0.8Zr0.2Oy催化剂中Ce3+占Ce3++Ce4+的摩尔分数为14.31%,比反应前高,可以推出Ce4+摩尔分数降低,这说明Ce4+作为活性组分参与到催化氧化反应中.同时,反应后的化学吸附氧占比降低,表明吸附氧参与催化反应,并在反应过程中被消耗.
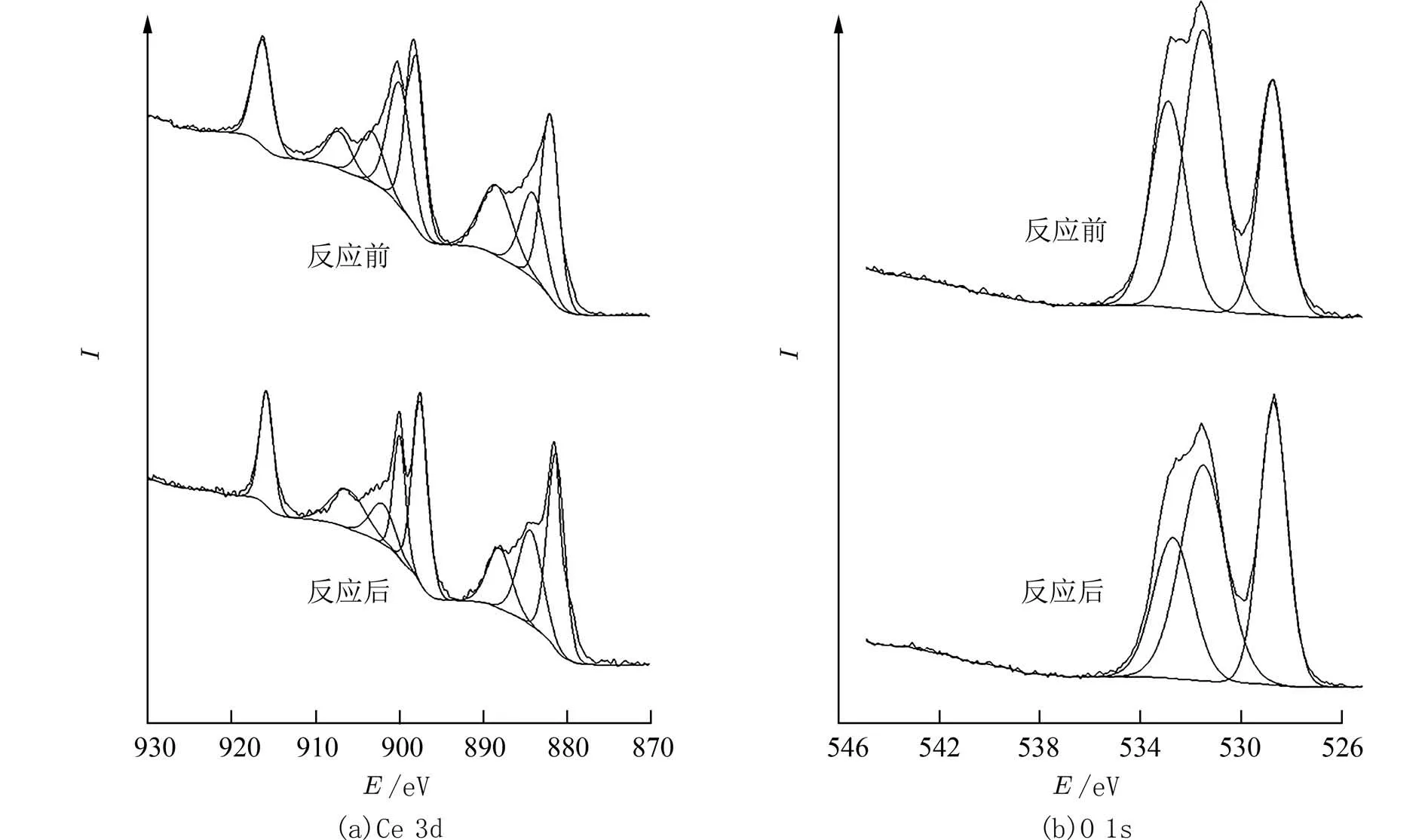
图6 Ce0.8Zr0.2Oy 反应前后的XPS 谱图Fig.6 XPS spectra of Ce0.8Zr0.2Oy before and after reaction

表3 CexZr1−xOy 的XPS 分析结果Tab.3 Results of the XPS analysis for CexZr1−xOy
2.5 n(Ce)/n(Zr)对甲醛去除率的影响CexZr1−xOy催化剂常温催化氧化去除甲醛的结果如图7 所示.CexZr1−xOy的甲醛降解率均高于CeO2、ZrO2.而Ce0.8Zr0.2Oy的甲醛催化活性又优于其他摩尔比的催化剂,其甲醛降解率于反应200 min 内保持峰值100%.而Ce0.6Zr0.4Oy、Ce0.4Zr0.6Oy、Ce0.2Zr0.8Oy催化剂在达到最高降解率分别为99.5%、98.86%、98.74%后,催化活性开始缓慢下降,在270 min 时分别降至86.5%、81.43%、75.32%.上述结果表明,n(Ce)/n(Zr)对催化剂的活性有显著影响,随着n(Ce)/n(Zr)的增加,催化剂的活性逐渐提高.说明Ce 的掺入大大提高了催化剂的催化活性;而Zr 的负载,显著提升了其氧化能力.
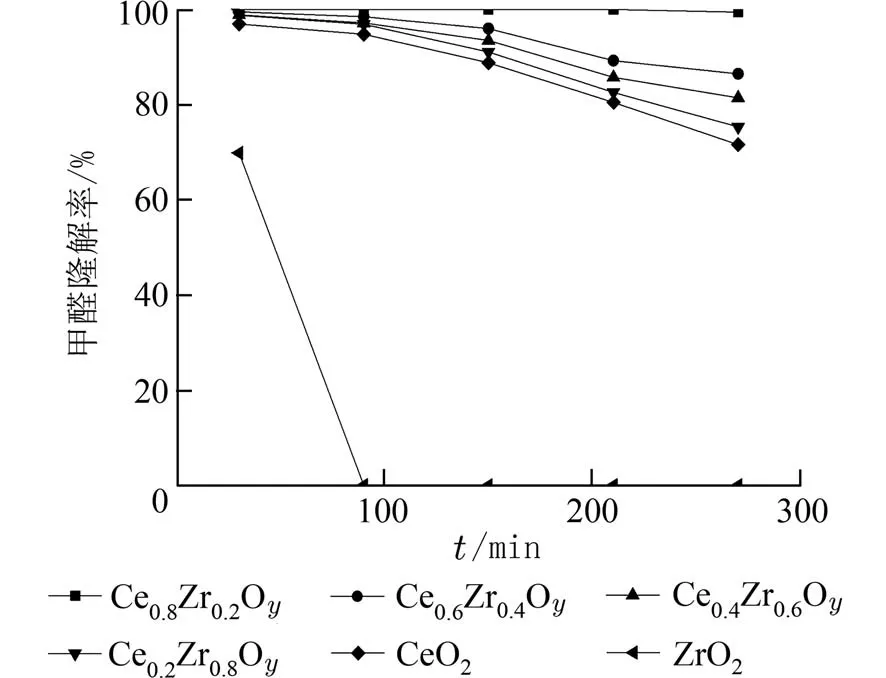
图7 甲醛降解率随时间变化曲线图Fig.7 Change curves of formaldehyde degradation rate over time
扫描电镜与N2吸附−脱附表征结果表明:Ce0.8Zr0.2Oy催化剂表面粗糙、疏松多孔,具有大的比表面积、孔体积与孔径,有利于气体在表面扩散与传质,让CH2O 更容易被吸附在活性组分表面从而被催化氧化.XPS 表征结果表明:Ce4+作为活性组分,将吸附的CH2O 氧化成CO2和H2O,同时自身被还原为Ce3+,而Ce3+又被活性氧物种氧化为Ce4+,补充活性组分.Zr4+参与轨道杂化改变材料的电子结构,使Ce4+表现出了更高的催化活性.因此,Ce0.8Zr0.2Oy催化剂催化活性高,此结论与实验结果一致.
2.6 Ce0.8Zr0.2Oy 催化性能为探究Ce0.8Zr0.2Oy催化性能,对其进行稳定性测试及焙烧再生测试,结果如图8 所示.在催化氧化实验进行了330 min时,甲醛去除率一直保持在97%以上.这归功于Ce0.8Zr0.2Oy催化剂拥有疏松多孔的形貌特征和丰富的反应位点.当反应进行了400 min 后,催化剂失活导致甲醛降解率开始明显降低,但依然保持在90%以上.直至催化氧化反应结束,Ce0.8Zr0.2Oy催化剂对甲醛的去除率保持在85%.
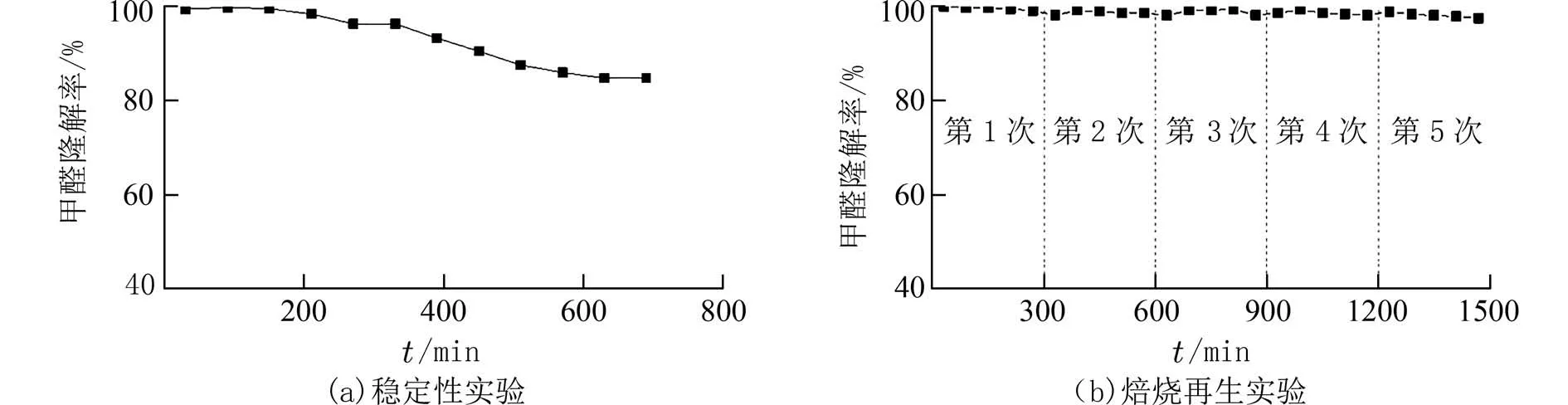
图8 Ce0.8Zr0.2Oy 催化性能测试图Fig.8 Catalytic performance test diagram of Ce0.8Zr0.2Oy
为考察Ce0.8Zr0.2Oy的再生能力,将经12 h 反应后失活的Ce0.8Zr0.2Oy收集起来,于500 ℃下焙烧4 h[29-30],重复5 次.图8(b)显示,第1 次反应结束时,甲醛降解率从100%降至98%.焙烧再生后进行第2 次反应,甲醛降解率又回升至100%.经过5 次的再生测试,Ce0.8Zr0.2Oy催化剂的甲醛降解率最终保持在97%.综上,Ce0.8Zr0.2Oy催化剂较稳定,具有良好的再生性.
为探究催化剂活性降低原因,利用傅里叶红外光谱仪对反应前后及再生前后的Ce0.8Zr0.2Oy进行测定.由图9 可见,反应后的Ce0.8Zr0.2Oy在3 384.56 cm−1处出现了吸收峰,此峰位于3 300 cm−1附近,峰宽而钝,为游离水的羟基吸收峰[31-32];而1 044.12 cm−1处的吸收峰为碳氧吸收峰[33].推测为催化剂将CH2O 催化氧化生成了H2O 和CO2.吸附在Ce0.8Zr0.2Oy表面的H2O 分解后以羟基形式存在;CO2进一步被转化成碳酸盐物质,而在反应后Ce0.8Zr0.2Oy电镜图(图10)中观察到的颗粒物正是这种物质.为证实上述推测,对反应后的Ce0.8Zr0.2Oy进行能谱分析.从图11 中得出,催化剂表面氧(O)、碳(C)和铈(Ce)的原子比例远高于其他元素.因此考虑,上述颗粒主要由O、C、Ce 元素组成.于是利用X 射线衍射仪对反应后的Ce0.8Zr0.2Oy进行测定,在图12 的XRD 结果中出现了Ce(CO3)2的衍射峰.焙烧再生后的Ce0.8Zr0.2Oy在1 076.34 cm−1处同样出现了碳氧吸收峰,电镜图中也观察到了少许颗粒物,说明Ce(CO3)2较稳定,难以被分解.
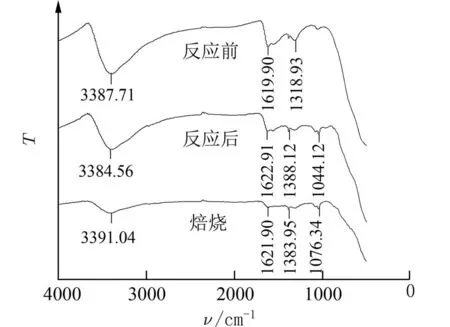
图9 Ce0.8Zr0.2Oy 红外图谱Fig.9 Infrared diagram of Ce0.8Zr0.2Oy

图10 Ce0.8Zr0.2Oy 扫描电镜图Fig.10 SEM image of Ce0.8Zr0.2Oy
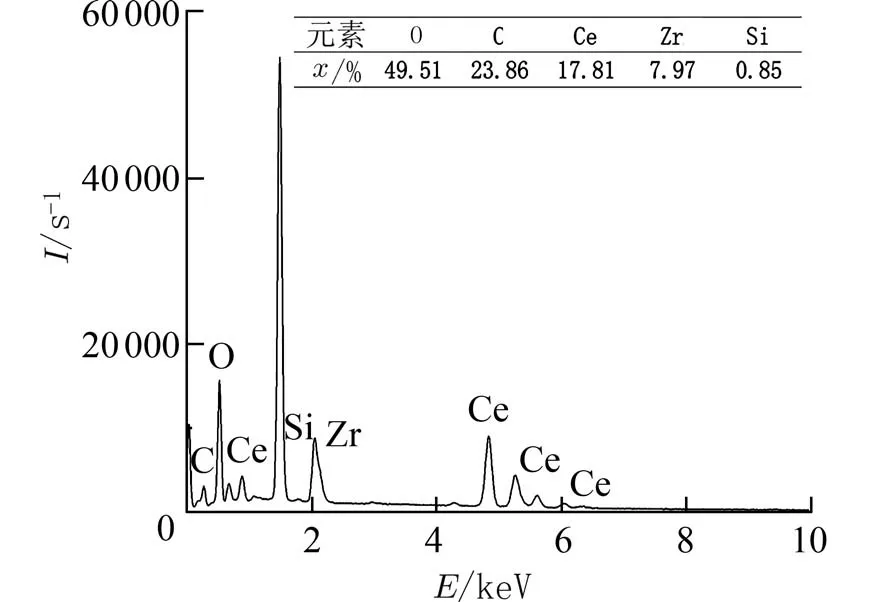
图11 Ce0.8Zr0.2Oy EDS 图谱Fig.11 EDS pattern of Ce0.8Zr0.2Oy

图12 Ce0.8Zr0.2Oy XRD 图谱Fig.12 XRD pattern of Ce0.8Zr0.2Oy
综上,在Ce0.8Zr0.2Oy催化氧化甲醛过程中,产物H2O 会与CH2O 竞争吸附在催化剂表面活性位点上,CH2O 吸附量减少导致催化剂活性降低,但升温即可恢复.而Ce(CO3)2的生成不仅消耗活性组分Ce4+,还占据催化活性位、堵塞孔道,升温也难以恢复,成为不可逆永久性失活[34].
综上所述,Ce0.8Zr0.2Oy催化氧化甲醛过程中,虽然因H2O 和Ce(CO3)2占据活性位点、堵塞孔道、消耗活性组分,导致催化剂失活、甲醛去除率降低,但反应12 h 后还能保持85%以上的去除率,较稳定.针对催化剂失活现象,对其进行焙烧再生处理,结果显示,5 次再生后甲醛去除率为97%,说明Ce0.8Zr0.2Oy有较好的再生能力.
3 结论
(1)常温除甲醛CexZr1−xOy催化剂通过共沉淀法制备.随着Ce 含量的增加,催化剂的催化活性逐渐提高.n(Ce)/n(Zr)为0.8/0.2,反应200 min 内保持100%的甲醛去除率.
(2)表征结果表明,Zr 成功地被掺杂到CeO2晶格中,形成铈锆固溶体.随着Ce 含量的增加,结构不规则的CexZr1−xOy催化剂的孔径、孔体积越来越大,比表面积也呈增加的趋势,有利于CH2O 在催化剂表面的扩散与传质.Ce0.8Zr0.2Oy催化剂存在更多活性组分Ce4+和化学吸附氧,为催化氧化甲醛提供反应位点.
(3)催化性能测试结果表明,反应12 h 后保持85%以上的甲醛去除率,5 次焙烧再生后去除率为97%,再生能力较好.催化剂失活原因为H2O 和Ce(CO3)2堵塞孔道、占据活性位点、消耗活性组分.
猜你喜欢
农业工程学报(2022年10期)2022-08-22
煤炭工程(2022年7期)2022-07-21
科学导报(2022年28期)2022-05-24
军事文摘(2021年24期)2022-01-11
安徽工业大学学报(自然科学版)(2021年3期)2021-09-08
装备维修技术(2020年5期)2020-11-20
舰船电子对抗(2019年5期)2019-12-04
发明与创新·大科技(2019年6期)2019-09-06
分析化学(2018年1期)2018-01-18
科技创新与应用(2017年35期)2017-12-19
