
Programmable endonuclease combined with isothermal polymerase amplification to selectively enrich for rare mutant allele fractions
2022-09-16 05:25JunmanChnTianQiuMihalMaukZhnSuYauanFanDnnisYuanQinhuaZhouYoulinQiaoHaimBauJianminYinJinzhaoSon
Chinese Chemical Letters 2022年8期
Junman Chn, Tian Qiu, Mihal G.Mauk, Zhn Su, Yauan Fan,Dnnis J.Yuan, Qinhua Zhou, Youlin Qiao, Haim H.Bau, Jianmin Yin,Jinzhao Son,,∗∗
a Key Laboratory of Clinical Laboratory Diagnostics (Ministry of Education), College of Laboratory Medicine, Chongqing Medical University, Chongqing 400016, China
b The Cancer Hospital of the University of Chinese Academy of Sciences (Zhejiang Cancer Hospital), Institute of Basic Medicine and Cancer (IBMC), Chinese Academy of Sciences, Hangzhou 310022, China
c Department of Mechanical Engineering and Applied Mechanics, University of Pennsylvania, Philadelphia, PA 19104, United States
d Department of Pathology, National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100021, China
e Center for Global Health, School of Population Medicine and Public Health, Chinese Academy of Medical Sciences and Peking Union Medical College,Beijing 100730, China
f Tianjin Key Laboratory of Lung Cancer Metastasis and Tumor Microenvironment, Tianjin Lung Cancer Institute, Tianjin Medical University General Hospital,Tianjin 300052, China
g Sichuan Lung Cancer Institute, Sichuan Lung Cancer Center, West China Hospital, Sichuan University, Chengdu 610041, China
ABSTRACT Liquid biopsy is a highly promising method for non-invasive detection of tumor-associated nucleic acid fragments in body fluids but is challenged by the low abundance of nucleic acids of clinical interest and their sequence homology with the vast background of nucleic acids from healthy cells.Recently,programmable endonucleases such as clustered regularly interspaced short palindromic repeats (CRISPR)associated protein (Cas) and prokaryotic Argonautes have been successfully used to remove background nucleic acids and enrich mutant allele fractions, enabling their detection with deep next generation sequencing (NGS).However, the enrichment level achievable with these assays is limited by futile binding events and off-target cleavage.To overcome these shortcomings, we conceived a new assay (Programmable Enzyme-Assisted Selective Exponential Amplification, PASEA) that combines the cleavage of wild type alleles with concurrent polymerase amplification.While PASEA increases the numbers of both wild type and mutant alleles, the numbers of mutant alleles increase at much greater rates, allowing PASEA to achieve an unprecedented level of selective enrichment of targeted alleles.By combining CRISPR-Cas9 based cleavage with recombinase polymerase amplification, we converted samples with 0.01% somatic mutant allele fractions (MAFs) to products with 70% MAFs in a single step within 20 min,enabling inexpensive, rapid genotyping with such as Sanger sequencers.Furthermore, PASEA’s extraordinary efficiency facilitates sensitive real-time detection of somatic mutant alleles at the point of care with custom designed Exo-RPA probes.Real-time PASEA’performance was proved equivalent to clinical amplification refractory mutation system (ARMS)-PCR and NGS when testing over hundred cancer patients’samples.This strategy has the potential to reduce the cost and time of cancer screening and genotyping,and to enable targeted therapies in resource-limited settings.
Keywords:Mutant allele enrichment Programmable endonuclease Liquid biopsy Mutation detection Point-of-care testing CRISPR-Cas9 Recombinase polymerase amplification Nucleic acid diagnostics
Somatic mutations are implicated in carcinogenesis, cancer progression, and therapeutic resistance.The detection of rare somatic mutant alleles (MAs) in cancer biopsy and liquid biopsy is challenged by their low abundance and sequence homology with a vast background of wild-type (WT) alleles from healthy cells.These challenges are greatly aggravated at early disease stages and during the evolution of drug resistant mutations.To detect low abundance somatic mutations, it is necessary to improve the signal to noise ratio by enriching the rare somatic mutant allele fractions (MAFs).This is accomplished by hybridizing nucleic acids of interest to nucleic acid probes in the presence of nucleic-acid guided endonucleases lacking catalytic activity (e.g., dCas9) [1,2]; by preferential enzymatic amplification of mutant alleles with specifically designed primers and DNA blockers [3–5]; by suppression of the amplification of WT alleles with capping nucleic acids [6]; by selective depletion of WT nucleic acids with RNase H at post-transcriptional level [7]; with restriction endonucleases [8,9]; and programmable endonuclease such as clustered regularly interspaced short palindromic repeats associated protein 9 (CRISPR-Cas9) [10,11] and Argnoautes [12–14], wherein unwanted background sequences are selectively removed from the sample.These various methods can be used independently or in combination.The targeted nucleic acids can then be detected either directly with probes during amplification, hybridization arrays, and sequencing.
Among the aforelisted methods, selective depletion methods based on programmable endonucleases [10–14] provide greater flexibility than those based on restriction endonuclease [8,9] since they do not rely on specific target sequences.Guided by synthetic oligonucleotides or silencing RNA, programmable endonucleases [15–18] remove the dominant, interfering (background) WT sequences to facilitate detection of scarce mutant alleles.While greatly improving the sensitivity of downstream genotyping methods, the efficacy of existing programmable Cas9-based enrichment assays is compromised by a significant fraction of unfertile binding events between enzyme and target, which due to the slow dissociation rate of Cas9 leave targets protected from cleavage [19] and by non-specific off-target cleavage that depletes precious biomarkers[11].To partially overcome these shortcomings, researchers have employed rounds of selective depletion of WT followed by polymerase chain reaction (PCR) amplification, enabling 10-fold enrichment of the fractions of mutant alleles [11].Despite these improvements, high sensitivity of mutant allele detection still requires the use of deep next generation sequencing (NGS) [11], rendering these methods laborious, time-consuming, and expensive.A single-step method that enriches the MAF to enable its detection by inexpensive and rapid means is highly desirable.
To address this need, we devised a new assay dubbed Programmable Enzyme-Assisted Selective Exponential Amplification(PASEA, Fig.1) that concurrently amplifies both WT and mutant alleles in the presence of guided endonuclease that targets only the WT allele.Given time, the variant that exhibits a superior trait(the mutant allele being less susceptible to cleavage) will dominate.PASEA requires temperature-matched polymerase and endonuclease.Herein, we use CRISPR-Cas9 programmed to cleave WT alleles in combination with isothermal recombinase polymerase amplification (RPA).We converted samples with 0.01% somatic MAFs to products with 70% MAFs in a single step (single pot)within 20 min, enabling inexpensive, rapid genotyping with such as Sanger sequencers.Previously, we reported the broad outlines of our approach [20].In this paper, we expound yet unpublished experimental data that demonstrates PASEA’s capabilities and its suitability for resource poor settings.Furthermore, we used PASEA to test 108 clinical tissue samples and 10 blood samples from cancer patients and compared PASEA with NGS and amplification refractory mutation system (ARMS)-PCR.Clinical samples were collected at the Cancer Hospital, Chinese Academy of Medical Sciences under the IRB-approved protocol (20/383-2579) (see Supporting information).
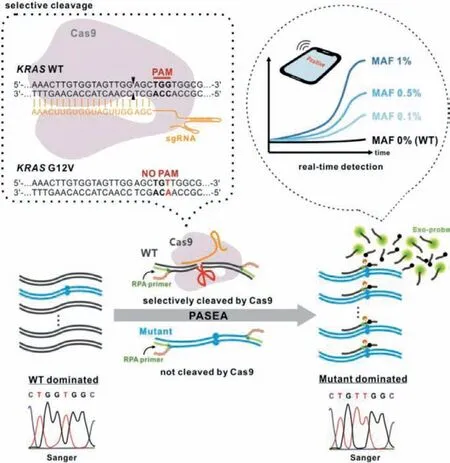
Fig.1.Principle of real-time PASEA.Directed by a single-stranded guided RNA(sgRNA), Cas9 selectively cleaves WT alleles with protospacer adjacent motif (PAM)site while sparing oncogenic mutation lacking PAM.The dotted frame illustrates WT and mutant allele sequences, the location of PAM site in the WT KRAS and its absence in KRAS G12.The sequences of the KRAS gene and mutants are from COSMIC online database (https://cancer.sanger.ac.uk/cosmic).Cleavage takes place between the third nucleotide and the fourth nucleotide upstream from the PAM site.While PASEA amplifies both WT and mutant alleles, the rate of amplification of mutant alleles far exceeds that of the WT, resulting in a product dominated by mutant alleles.Exo-probe indicates the number of amplicons and enables quantification in real time.
PASEA relies on selective cleavage to obtain much greater amplification rates of mutant alleles than of WT alleles.The contrast between the PASEA and RPA [in the absence of Cas9 and sgRNA ribonucleoprotein (RNP)] amplification rates of WT genomic DNA is striking (Fig.2a).Within 10 min, RPA produced about 109amplicons while PASEA produced less than 105—four orders of magnitude less.When PASEA acts on a standardKRASG12V (MAF 5%)sample (Fig.2a), the numbers of bothKRASG12V and WTKRASamplicons increase as time increases but theKRASG12V amplifies at a much greater rate than WTKRAS.Due to selective amplification, the number of amplicons of WTKRASin the blend should be much less than the number of amplicons when PASEA is applied to pure WTKRAS(blue bars).After ∼3 min, the products of the standard sample (5% MAF) are dominated by the mutant allele (green bars).
To verify that the amplicons are, indeed,KRASG12V, we subjected the PASEA products of 60 ng genomic DNA (gDNA),initially with 5%KRASG12V to Sanger sequencing (Fig.2b-i)and estimated the MAFs with the Mutation Surveyor Software(https://softgenetics.com/mutationSurveyor.php) (Fig.2b-ii).Consistent with our quantitative real-time PCR (qPCR) results, the Sanger sequencing data show that the MAF has increased from 5% to 70% after 3 min PASEA incubation and to nearly 100% after 5 min or longer incubation.PASEA provides highly efficient enrichment with RNP concentrations ranging from 0.1 μmol/L to 1 μmol/L RNP (Fig.S2 in Supporting information).
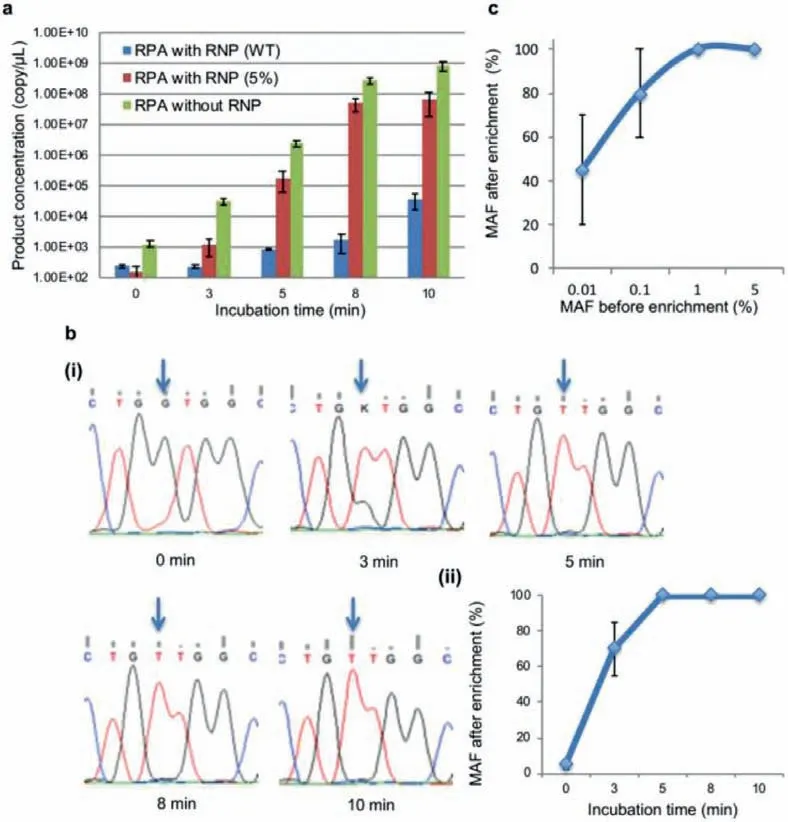
Fig.2.PASEA exponentially enriches MAF.(a) Pure WT-KRAS dsDNA and blends containing KRAS G12V dsDNA (MAF 5%) were subjected to PASEA (0.1 μmol/L RNP) for various time spans along with a control group subjected to RPA without RNP.The number of amplicons is inferred from the threshold time of pre-calibrated qPCR (Fig.S1 in Supporting information). n = 3.(b) Blends sample containing KRAS G12V dsDNA (MAF 5%) were subjected to PASEA (0.1 μmol/L) for various time spans and then evaluated by Sanger sequencing: (i) sequencing results of PASEA products; (ii) estimated MAF as a function of PASEA incubation time.(c) Average MAF following PASEA incubation(enrichment).The MAF of each sample (total gDNA, 60 ng) before enrichment is from 0.01% to 5%.The PASEA incubation time is 20 min with 0.1 μmol/L RNP.
The minimal required incubation time depends on the sample’s MAF (Fig.S3 in Supporting information).When MAF = 1%, incubation time of 10 min suffices to deplete the WT to undetectable level in the Sanger sensorgram.When MAF = 0.1%, 10 min PASEA enriches the MAF to approximately 60%, and 20 min PASEA makes mutant allele fraction nearly 100%.Hence, in our subsequent experiments, we used 20 min incubation time.Longer incubation times such as 30 min result in noisy sensorgrams possibly due to the presence of spurious amplicons.
To evaluate PASEA’s sensitivity, we subjected a standard genomic DNA panel with 0%, 0.01%, 0.1%, 1% and 5%KRASG12V MAFs to 20 min PASEA incubation and examined the incubation products with Sanger sequencing (Fig.2c).Sanger sequencing identified the presence ofKRASG12V in all the PASEA products of the samples with MAF = 0.1%, 1%, or 5% (n= 3).According to Sanger results,PASEA increased the MAFs to nearly 100% in the 5% (20-fold enrichment) and 1% (100-fold enrichment) samples; to 80% (800-fold enrichment) in the 0.1% samples.Seven out of 10 PASEA products of samples with initial MAF = 0.01% (n= 10), which equals to 2 copies mutant alleles in 60 ng total gDNA, were identified positive by Sanger, with the products’MAF averaging at 40% (4000-fold enrichment).The 3/10 negatives are attributable to a sampling error.Statistically, it is reasonable to expect that mutant alleles were completely absent in the “false negative” samples.Importantly, PASEA did not produce any false positives.
Previous researchers [10] used Cas9-based cleaving assay(DASH) without concurrent amplification.How does PASEA compare with DASH? A 20 min incubation with DASH enabled detection of only MAF>1% in our hands (Fig.S4 in Supporting information) and MAF>0.1% under optimal conditions (Table S2 in Supporting information) [10] while PASEA facilitated detection of 0.01%with downstream Sanger sequencing.PASEA has about two orders of magnitude better performance than DASH in terms of detectable MAF.
PASEA followed with Sanger sequencing and/or qPCR provides sensitive, two-stage detection of rare alleles.To meet the needs of resource poor settings, we designed a single stage, closed pot assay.Our real-time assay uses an Exo-RPA probe (Fig.S5a in Supporting information) comprised of an abasic nucleotide analogue(tetrahydrofuran residue, THF) with a flanking dT-fluorophore at one side, a dT-quencher on the other, and a C3-spacer to block polymerase extension.When free in solution, the probe’s fluorophore is quenched by the quencher located 2–5 bases away from the fluorophore.The THF provides a substrate for the exonuclease III enzyme (included in the TwistAmpR○exo kit) when the probe hybridizes with the amplicon to form a double-strand context.Enzymatic digestion of the THF separates the fluorophore from the quencher, resulting in fluorescent emission.
The short template sequence (∼160 bp) [21,22] of the cell-free DNA challenges probe design.It is difficult, if not impossible, to avoid an overlap between the probe’s hybridization site and the sgRNA protospacer sequence.We evaluated several Exo-RPA probe sequences (Fig.S6 in Supporting information) and selected the best performer (Fig.S5b in Supporting information) that hybridizes to the amplicon’s middle region.Since the THF localizes to theKRASG12V/D location, the Exo-RPA probe does not discriminate between the WT and mutant alleles, but instead reports on the total number of amplicons (Fig.S5c in Supporting information).
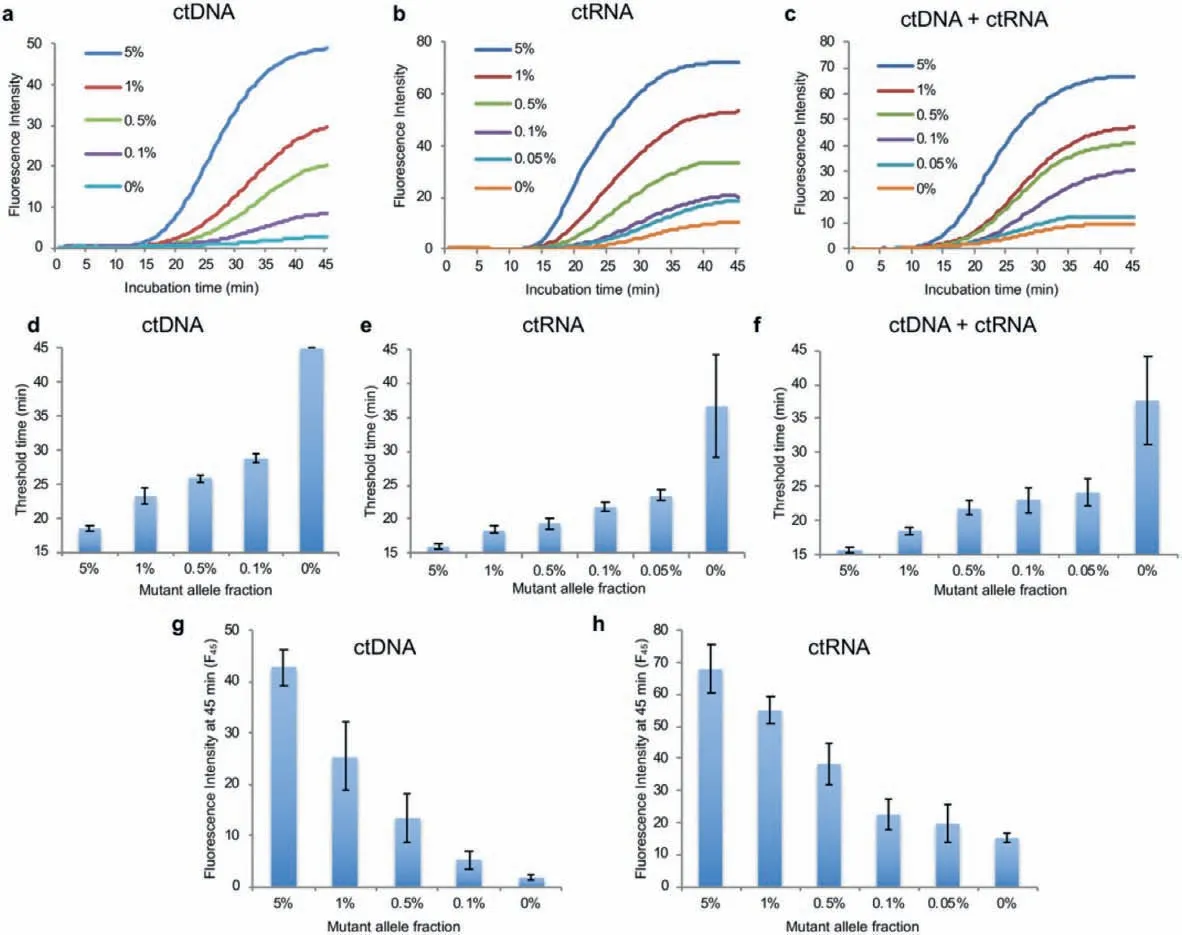
Fig.3.Real-time PASEA performance.Amplification curves of cfDNA (a), RNA (b), and mixture of cfDNA and RNA (c) with different MAF.Threshold time as a function of initial MAF when detecting cfDNA (d); RNA (e); and a mixture of cfDNA and RNA (f).Fluorescence intensity at 45 min as a function of MAF when detecting cfDNA (g) and RNA (h).
Since the probe and the sgRNA have overlapping sequences, the probe affects Cas9 cleaving efficiency (Fig.S7 in Supporting information).At probe concentrations of 120 and 240 nmol/L, real-time PASEA discriminates well between 5%KRASG12V and WT alleles while at higher probe concentrations (e.g., 600 nmol/L), there is little contrast between 5%KRASG12V and WT alleles; likely because of probe interference with the sgRNA hybridization.In all our subsequent experiments, we used 240 nmol/L probe concentration that provided a brighter signal than the 120 nmol/L concentration, therefore reducing demands on the signal detector.
To address the interference between the probe and the RNP, we examined the effect of RNP concentration on the real-time amplification curve in the presence of 240 μmol/L probe (Fig.S8 in Supporting information).Reaction mixes with 0.1 (a) and 0.08 μmol/L(b) RNP discriminated well between samples of 0% and 5%KRASG12V gDNA while 0.05 μmol/L RNP (c) provided less satisfactory contrast.To further finetune our assay for cell free DNA (cfDNA)detection, we compared the effect of RNP concentrations on realtime PASEA acting on a standardKRASG12D cfDNA control (0%,0.1%, 1% and 5% MAF).The assay with 0.08 μmol/L RNP rendered 0.1% MAF (f) detectable while the same MAF was not detectable with 0.1 μmol/L RNP.
At early disease stages, cell free mutant alleles are present in body fluids at very low concentrations.Cell-free RNA, predominantly comprised of small RNAs and mRNAs, is present in peripheral blood, partly protected from degradation by its packaging into exosomes [23,24].To increase the number of templates (biomarkers) for our assay, we target cell free, tumor derived fragments of both DNA and RNA [25].Since the cfDNA (∼160 nt) and theKRASexon 2 (122 nt) share a short common sequence (125 nt, Fig.S9a in Supporting information), we designed and tested various primers for short templates to concurrently amplify both ctDNA and complementary DNA (Fig.S9 and Table S1 in Supporting information).We then selected the primer set that provides the shortest threshold time.We targetedKRASG12D, which likeKRASG12V, lacks the “NGG” protospacer adjacent motif (PAM) sequence.Real-time PASEA with primers RPA-ct-Fw-1/RPA-ct-Rv-2 detectsKRASG12D in 20 ng of standard cfDNA in the absence of reverse transcriptase (RT) (Figs.3a and d); in 400 ng of purified mRNA (∼120,000 copies) in the presence of RT (Figs.3b and e); and in a mixture of 10 ng cfDNA and 200 ng mRNA in the presence of RT (Figs.3c and f) at various MAFs.The quantities of nucleic acid used in our experiments are comparable with those in patient samples.As expected, the threshold time (defined as the time until signal intensity exceeds 10% of signal saturation level) increases as the MAF decreases (Figs.3d–f).PASEA readily detects 0.1% MAF cfDNA (∼6 copies in 20 ng, Figs.3a and d), 0.05% G12D mRNA (∼60 copies in 400 ng, Figs.3b and e) and 0.05% mixture of cfDNA and mRNA(Figs.3c and f).
Interestingly, the fluorescence intensity of the amplification curve’s plateau increases as the MAF increases.The fluorescence intensity of the plateau at 45 min (F45) correlates well with the MAF (Figs.3g and h) and with the threshold time (Fig.S10 in Supporting information), providing yet another metric for semiquantitative estimation of the MAF.
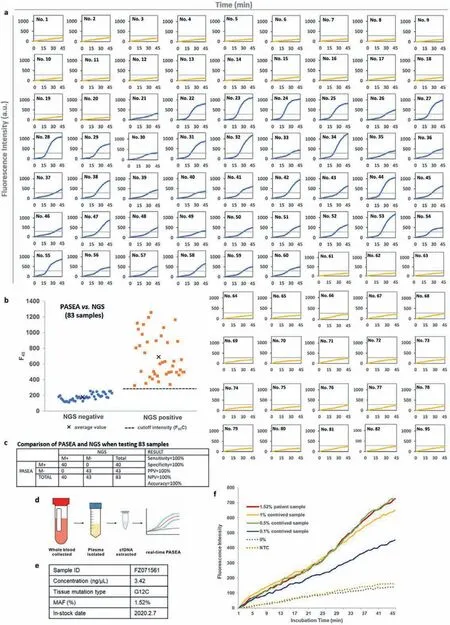
Fig.4.Real-time PASEA successfully detects gDNA and cfDNA in clinical samples.(a) Real-time PASEA amplification curves of 83 samples that were also tested with NGS.The amplification curves of the 40 positive samples (Nos.21–60) are in blue and the 43 negative samples (Nos.1–20, 61–82, 95) are in yellow.(b, c) F45 values of patient samples(83) compared with tissue NGS genotyping (“gold standard”).The dashed horizontal line and the symbol “x” denote, respectively, the F45 cutoff value and the average F45 value.(d) Workflow of blood testing with real-time PASEA.(e) Details of the NGS KRAS positive blood sample.(f) Real-time PASEA amplification curves of cfDNA samples with various mutant allele fractions.NTC, non-target control.
Is PASEA applicable to clinical samples? We collected 108 tissue samples from colorectal cancer (62/108), pancreatic cancer (1/108),and lung cancer patients (45/108) by either resection or biopsy at the Cancer Hospital of the Chinese Academy of Medical Sciences(Beijing, China).gDNA was extracted from these tissue samples and tested with clinical NGS (83 samples) and ARMS-PCR (97 samples).Forty samples were positive toKRASmutations (G12V, D, S,C and R) with MAF ranging from 1% to 39%.68 samples were negative forKRASmutations (Table S3 in Supporting information).The extracted gDNAs were diluted to 10 ng/μL and then tested with our real-time PASEA.Real-time PASEA was deemed positive when F45exceeded the cutoff (F45C), defined as the average F45plus 3 standard deviation (SD) (95% confidence level) for standard WT gDNA at 20 ng/μL, which is greater than the DNA concentration in our clinical samples (∼10 ng/μL) (Table S3).Real-time PASEA correctly identified 40/40 samples as positive and 68/68 samples as negative, exhibiting 100% sensitivity, specificity, positive predictive value, negative predictive value, and concordance with NGS(Figs.4a–c) and/or ARMS-PCR (Fig.S11 in Supporting information)genotyping forKRASG12 mutations.In all cases, the emission intensity (F45) correlated well with the threshold time (Fig.S12 in Supporting information).
Plasma samples were collected from 10 lung cancer patients and banked at −80 °C (Fig.4d).NGS identified only one sample as positive forKRASmutation (KRASG12C, MAF = 1.52%, Fig.4e).To compensate for the positive clinical samples’scarcity, we spiked Horizon standard cfDNA (WT) controls into the positive sample to form contrived samples with MAFs of 1%, 0.5% and 0.1%.The 10 patient samples and the 3 contrived samples were subjected to realtime PASEA.The amplification curves of positive samples clearly differentiated from the curve of the WT control (0%) (Fig.4f), indicating that real-time PASEA detects ctDNA in blood samples with high sensitivity.
Point-of-Care analysis of nucleic acids for cancer profiling and infectious disease diagnosis is highly desirable [26,27].Real-time PASEA is relatively simple to carry out, does not require strict temperature control, and can be used at the point of care [28].We implemented real-time PASEA in our multifunctional isothermal amplification microfluidic (MIAR) chip [28] that extracts and concentrates nucleic acids from a sample and mates with a homemade portable isothermal amplification processor (Fig.S13a in Supporting information) [29].We carried out real-time PASEA on samples comprised of various concentrations of standardKRASWT cfDNA and G12D ctDNA spiked in phosphate buffered saline (PBS).Samples with MAF of 0.5% ofKRASG12D were readily detected within 40 min (Figs.S13b and c in Supporting information).The lengthy incubation time is partially due to the slow temperature ramp of our homemade incubator.Real-time PASEA exhibited detection limit of about 87 copies when operating with samples comprised of 60 ng cfDNA.This can likely be improved further with additional optimization.
Researchers have identified various oncogenic mutations responsible for the initiation and maintenance of cancer and the mechanisms of resistance to targeted therapeutics [30] Methods for cost effective, non-invasive cancer genotyping are needed to enable targeted therapies.An attractive genotyping method relies on identifying tumor derived, aberrant nucleic acids in body fluids (liquid biopsy).However, the identification of tumor-associated nucleic acid fragments in body fluids is challenged by their low abundance and sequence homology with the vast background of nucleic acids from healthy cells.
Among the current programmable endonuclease-based mutant allele enrichment methods [10–13], the CRISPR-mediated, ultrasensitive detection of target DNA by PCR (CUT-PCR) [11] is a promising assay to enrich mutant alleles’fraction by 2–3 rounds of selective depletion of WT and subsequent PCR.CUT-PCR has successfully increased MAF by 27-fold in most samples that contained over 0.1%MAF before cleavage [11].In combination with deep sequencing,CUT-PCR enables detection of 0.01% mutant alleles.
There are, however, a few factors that limit the enrichment level achievable with assays such as CUT-PCR.Although Cas9 preferentially cleaves WT alleles, it also cleaves, albeit, to a lesser degree, off-target mutant alleles.While off-target and target cleavage rates depend on the guide RNA design, Cas9 variant, and assay conditions, samples with low MAF (e.g., 0.01%) contain just a handful of molecules of clinical interest and any loss of critical biomarkers compromises assay’s sensitivity.Furthermore, not all guide–protein-target triplexes are productive.By some estimates, fewer than 90% triplexes are cleaved [31].Since the dissociation rate of the triplex is very slow [19], a significant fraction of WT DNA is protected from cleavage but amenable to PCR amplification.For example, if an assay cleaves only 90% of the WT alleles, the MAF can be enriched by less than 10-fold in a single step.
Herein, we propose a simple remedy that combines cleavage with concurrent polymerase amplification to overcome the shortcomings of the cleavage only assays.Our assay amplifies concurrently mutant alleles of interest and WT alleles in the presence of relatively high concentration of guided endonuclease.While the copy numbers of both the WT and mutant allele increase with time, the latter do so at much greater rate, alleviating any concerns of losing valuable biomarkers.Compared to restriction site mutation assay [8,9], PASEA has less limitation of target sequence.
PASEA’s very high enrichment capability enables libraries preparation for rapid, low-cost sequencers such as Sanger.It also offers the opportunity for real-time detection of mutant alleles in a closed pot without a sequencer, eliminating the need to open amplicon-rich tubes and risking the contamination of the workspace.Clinical evaluation of real-time PASEA exhibits good concordance with NGS and ARMS-PCR genotyping forKRASG12 mutations when testing tissue samples.Real-time PASEA has successfully identified the presence of mutant alleles in all positive samples and yielded no false positives in liquid biopsy.Real-time PASEA enables unprecedented level of enrichment and detection with relatively simple instruments, providing effective means for cancer screening and targeted therapies in low resource settings.
Although real-time PASEA’s reliance on Cas9 limits its use to sequences in which the PAM motif is present in the WT allele and absent in the mutant allele, the “NGG” PAM site is shorter and appears more frequently than restriction endonuclease recognition sites giving PASEA advantage over restriction-based assays.Moreover, Leeet al.[11] estimate, that with the use of various orthologonal CRISPR endonucleases such as SpCas9 and FnCpf1, Cas9-like proteins can target about 80% of known cancer-linked substitution mutations registered in the Catalogue of Somatic Mutations in the Cancer (COSMIC) database.Furthermore, our two-stage PASEA and our real-time PASEA can be extended to use other families of endonuclease such as Argonaute proteins [32] that do not require the presence of PAM and therefore are more versatile.PASEA’s simplicity makes it amenable for use in resource-limited settings with potential significant impact on global health and in applications other than cancer.
Declaration of competing interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: The University of Pennsylvania has filed for a patent for the method described in this paper with Jinzhao Song, Junman Chen,and Haim H.Bau listed as co-inventors.
Acknowledgments
This work was supported by China Scholarship Council, NIH grant to the University of Pennsylvania (No.K01 1K01TW011190–01A1), NIH grant to the University of Pennsylvania (No.R21 CA228614–01A1), and Beijing Hope Run Special Fund from the Cancer Foundation of China (Nos.LC2019L04 and LC2020A36).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.11.065.
 Chinese Chemical Letters2022年8期
Chinese Chemical Letters2022年8期
- Chinese Chemical Letters的其它文章
- Adsorptive removal of PPCPs from aqueous solution using carbon-based composites: A review
- A review on hollow fiber membrane module towards high separation efficiency: Process modeling in fouling perspective
- Recent advances in DNA glycosylase assays
- Chiral pillar[n]arenes: Conformation inversion, material preparation and applications
- Recent progress in carbon-based materials boosting electrochemical water splitting
- Working principle and application of photocatalytic optical fibers for the degradation and conversion of gaseous pollutants