
Recent advances in DNA glycosylase assays
2022-09-16 05:23LiliWangHuigeZhangWeiChenHongliChenJianxiXiaoXingguoChen
Chinese Chemical Letters 2022年8期
Lili Wang, Huige Zhang, Wei Chen, Hongli Chen, Jianxi Xiao, Xingguo Chen
State Key Laboratory of Applied Organic Chemistry, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, China
ABSTRACT Genomic deoxyribonucleic acid (DNA) is selected as the ideal carrier for preserving and transmitting the genetic information over the course of evolution.However, the genomic DNA is constantly exposed to various endogenous and environmental threats, causing a diversity of damaged bases, lesions, mismatches and base-pair modifications in the genome, eventually leading to genomic instability and cancers.Base excision repair (BER) is the most important repair mechanism, repairing a variety of DNA damages arising from oxidation, alkylation, methylation, deamination, and hydrolysis reactions.DNA glycosylases are responsible for initiating the first step of the BER pathway through cleaving the N-glycosidic bond between the damaged base and the DNA backbone.However, abnormal DNA glycosylases are associated with a variety of diseases such as cancer, cardiovascular disease, neurological disease and inflammation, suggesting the important role of DNA glycosylases in cancer diagnosis and treatment.Therefore, it is highly desirable to monitor the activity of DNA glycosylases, gaining a deep understanding of the restoration process of damaged DNA and clinical diagnosis.Recently, a series of novel DNA glycosylases detection methods with excellent performance have been developed.In this minireview, we summarize the recent advances in DNA glycosylase assays including amplification-free assay and amplification-assisted assay.Firstly, a brief introduction of amplification-free assay for DNA glycosylase is given.Then, amplification-assisted assays for DNA glycosylases are discussed in detail.Ultimately, the conclusion and prospects of the directions of DNA glycosylase assays are provided.
Keywords:DNA glycosylases Base excision repair Polymerase chain reaction Isothermal signal amplification Nanomaterial-based biosensors
1.Introduction
Deoxyribonucleic acid (DNA) is one of the most important biological macromolecules in living organisms, and almost all of the DNA is double-helix structure by pairing adenine (A) with thymine(T) and guanine (G) with cytosine (C) according to Watson-Crick pairing rules [1].Based on the intrinsic stability of double-helix structure, DNA is selected as the ideal molecule for coding and storing the genetic information, and it can be remarkably resistant to chemical attack by solvent and exogenous chemical agents [2].Despite its high fidelity rate, its stability with respect to ensuring the preservation of its coding content is limited because it continually suffers assault from exogenous and endogenous agents accounting for thousands of lesions per mammalian genome per day,destabilizing the duplex thermodynamically relative to the corresponding undamaged parent DNA duplex and leading to a wide variety of DNA modifications [3].These modifications may cause mutations in transcribed RNA and replicated DNA, altering the ability of regulatory elements to be recognized by DNA binding proteins,leading to cell death by blocking transcription and replication [4,5].
The nucleobases of DNA in the cells of all organisms are subject to constant genotoxic bombardment, both exogenous (e.g.,ultraviolet and ionizing radiation, chemical combustion products and relatively high temperature) and endogenous (e.g., reactive oxygen species, nucleases and alkylating agents) damage [6].A variety of lesions, such as DNA base lesions, single-strand breaks, and full double-strand breaks, can occur to most parts of the DNA structure [7].Chemical modifications are the most common lesions,and there are four major classes of base lesions including oxidation, deamination, alkylation, and hydrolysis [8].Reactive oxygen species (ROS) including hydrogen peroxide, hydroxyl radical and superoxide anion can lead to formation of oxidative lesions [9].The most abundant oxidative lesions of ROS is the oxidation of G to 8-oxoguanine (8-oxoG), resulting in transversion mutations from A/T to C/G or G/C to T/A [10].Deamination is the replacement of an N atom with an O atom, especially of exocyclic amines[11], and the deamination of the exocyclic amine of A and G produces hypoxanthine (I) and xanthine.Alkylation is one of the less common types of base lesions, but they are often the most mutagenic [12].Both pyrimidines and purines can be methylated to methylpyrimidines and methylpurines by adding methyl groups to any available amine.Hydrolysis may occur spontaneously in theNglycosidic bond to generate an abasic (apyrimidinic/apurinic, or AP)siteviathe action of a DNAN-glycosylase.
In order to maintain genome integrity and a low rate of mutation frequency, DNA damage must be corrected efficiently.Base excision repair (BER) is one of the central DNA repair pathways, and it can counter the mutagenic and cytotoxic effects of DNA damage, playing a critical role in maintaining genomic integrity [13].The BER pathway efficiently corrects most non-bulky DNA base lesions and repairs the vast majority of lesions that occur in DNA[14].BER generally invokes five following steps [13,14]: (i) Excision of the damaged nucleobases by a DNA glycosylase to create an AP site; (ii) Cleavage of the phosphodiester bond by an AP endonuclease; (iii) Removal of the resulting termini; (iv) DNA synthesis to replace the damaged nucleotide(s) by a DNA polymerase; and (v)ligation of the residual nick.
As one of the most important repair enzymes, DNA glycosylases are responsible for initiating the first step of the BER pathway through cleaving theN-glycosidic bond between the damaged base and the DNA backbone [15].DNA glycosylases may be broadly classified as monofunctional or bifunctional according to the reaction mechanisms [16].Monofunctional DNA glycosylases can hydrolyzeN-glycosidic bonds to create a common AP site which can be cleaved by an AP endonuclease.Bifunctional DNA glycosylases have both glycosylase and lyase activities, and they are already equipped with lyase capability to nick the AP site after removing incorrect bases.
The aberrant level of DNA glycosylase in human cells may cause the malfunction of BER and eventually various diseases including aging [17–19], cancer [20–23], neurodegeneration [24–27].Therefore, it is highly desirable to monitor the activity of DNA glycosylases, gaining a deep understanding of the restoration process of damaged DNA and clinical diagnosis.
Traditional methods for the detection of DNA glycosylase are mainly based on gel electrophoresis (GE) [28,29], mass spectrometry (MS) [30,31], high-performance liquid chromatography (HPLC)[32], and enzyme-linked immunosorbent assays (ELISA) [33].However, these methods usually suffer from poor sensitivity and laborious procedures.Besides, GE involves hazardous radiation, HPLC and MS are usually high background signals because the bases are artificially damaged in the process of sample collection and preparation, while ELISA may underestimate the actual damaged base level owing to the loss of samples during multi-step washing.
In recent years, a variety of emerging technologies have been developed for the detection of DNA glycosylase activity, such as colorimetry [34–36], fluorometry [37–39] and electrochemiluminescence (ECL) [40].Although previous review article explored the variety of DNA glycosylase assays, it mainly focused on the sources of DNA lesions, their repair and the emerging detection methods for partial DNA glycosylases [6].In this mini-review, we summarize the recent advances in DNA glycosylase assays including amplification-free assay and amplification-assisted assay (Fig.1).Firstly, a brief introduction of amplification-free assay for DNA glycosylase is given.Then, the detailed discussion is amplificationassisted assay including polymerase chain reaction (PCR), isothermal amplification and nanomaterial-based signal amplification for DNA glycosylase.Ultimately, the conclusion and prospects of the directions of DNA glycosylase assays are provided.
2.Amplification-free assay for DNA glycosylase
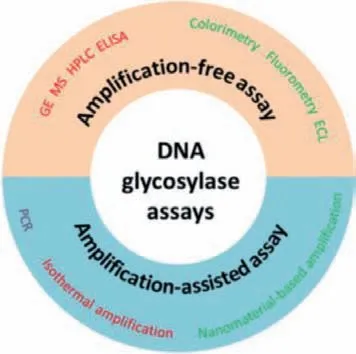
Fig.1.Schematic illustration of DNA glycosylase assay.
In order to overcome some drawbacks of traditional detection methods for DNA glycosylase, some amplification-free assays for DNA glycosylase such as colorimetric, fluorometric, and ECL assays are being developed to improve convenience and sensitivity.The colorimetric method possesses the advantages of convenience and being cost-effective, and its signal can be directly monitored by the naked eye.As shown in Fig.2A, Jianget al.[34] reported a colorimetric method for DNA glycosylase activity assay with the assistance of lambda exonuclease cleavage.Human 8-oxoguanine DNA glycosylase 1 (hOGG1) can selectively cleave the DNA duplex containing an 8-oxoG, generating a new DNA duplex with a recessed 5′-PO4terminus which can be digested by lambda exonuclease, releasing the free G-quadruplex single strand.The released G-quadruplex can bind with hemin to form a catalytically active G-quadruplex-hemin DNAzyme which can catalyze the H2O2-mediated oxidation of ABTS2−to the colored ABTS−and then the activity of hOGG1 could be indicated by the UV-vis absorption intensity.The fluorescence analysis method has been widely applied in detecting DNA glycosylase.As shown in Fig.2B, Liuet al.[37] developed a fluorescence method for DNA glycosylase activity assay using stem-loop molecular beacons with fluorescence resonance energy transfer (FRET).The molecular beacon modifying with two uracil bases in the stem are used as the substrate of uracil-DNA glycosylase (UDG).In the presence of UDG, the uracil bases in the substrate can be cleaved to produce AP sites, and then the melting temperature of the molecular beacon could be decreased significantly due to the non-complementary DNA strands.As a result, fluorophore-labeling beacons are dissociated readily from complementary strand, and consequently a significantly enhanced fluorescence signal is generated in response to UDG.ECL combines the advantages of luminescence (e.g., high sensitivity and wide dynamic range) and electrochemical (EC) techniques(e.g., simplicity, stability, and facility) [41,42].As shown in Fig.2C,Guoet al.[40] designed an ECL sensor for DNA glycosylase activity assay employing a novel spermine conjugated ruthenium tris-(bipyridine) derivative (spermine-Ru) which binds specifically with 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxodGuo).In the sensor,double-stranded DNA (dsDNA) film containing 8-oxodGuo was immobilized on a gold electrode by self-assembly.The ECL signal intensity was found to correlate with the amount of 8-oxodGuo on the surface.However, the 8-oxodGuo can be cleaved and repaired in the presence of formamidopyrimidine-DNA glycosylase (FPG),preventing the binding of spermine-Ru with 8-oxodGuo, resulting in the decrease of the ECL intensity.
3.Amplification-assisted assay for DNA glycosylase
3.1.Polymerase chain reaction (PCR)
PCR is the most popular amplification strategy developed by Kary Mullis [43].The PCR processes employ denaturation, annealing, and subsequent extension with expensive thermal cyclers.Billions of copies from a single target molecule can be generated after running about thirty repeating cycles by PCR thermal cycler.Re-
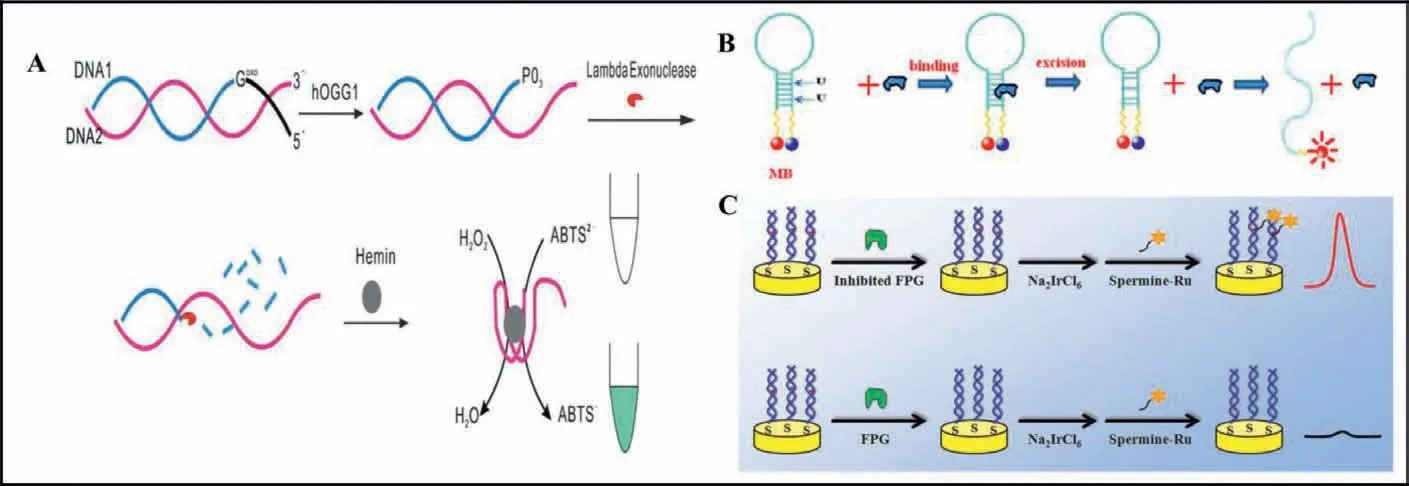
Fig.2.Schematic illustration of amplification-free DNA glycosylase assay.(A) A colorimetric method for hOGG1 activity assay.Copied with permission [34].Copyright 2013,Royal Society of Chemistry.(B) A fluorescence method for UDG activity assay.Copied with permission [37].Copyright 2014, Elsevier.(C) An ECL sensor for FPG activity assay.Copied with permission [40].Copyright 2015, Elsevier.
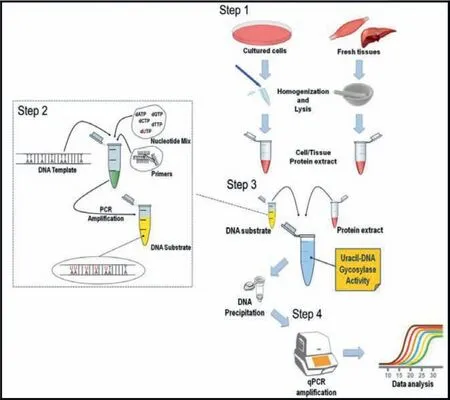
Fig.3.Schematic illustration of amplification-assisted DNA glycosylase assay with qPCR.Copied with permission [44].Copyright 2019, Springer Link.
cently, PCR has been used to detect DNA glycosylase based on its huge benefits of high sensitivity.As shown in Fig.3, Galderisiet al.[44] proposed a quantitative assay for UDG activity by PCR.Firstly,protein extracts including UDG were obtained from cell cultures and fresh tissues.Then, the DNA substrates containing deoxyuridine triphosphates (dUTPs) were incubated with UDG of cell/tissue extract, and the damaged uracil would be cleaved and repaired,consequently the UDG activity is readily derived from the quantitative PCR (qPCR) amplicon yield.This method allows the absolute quantification of the undigested amplicons in the samples treated with different protein extracts.However, PCR requires a complex and expensive thermal cycler for mediating denaturation, annealing, and subsequent extension, which largely limits the application of PCR in resource-limited settings.
3.2.Isothermal amplification methods
Isothermal amplification offers an alternative to PCR, allowing for nucleic acid amplification to occur at a single and constant temperature, eliminating the need for a thermocycler.Isothermal amplification has already become a powerful tool for efficiently detection of DNA glycosylase, such as target-recycling amplification with endonuclease, exonuclease and polymerase, strand displacement amplification (SDA), exponential amplification reaction (EXPAR), rolling-circle amplification (RCA), loop-mediated amplification (LAMP), and enzyme-free strand displacement reactions (SDR).
3.2.1.Target-recycling signal amplification with exonuclease,endonuclease and polymerase
T7 exonuclease (T7 exo) can catalyze the removal of 5′-mononucleotides from the 5′-termini of dsDNA without degradation of single-stranded DNA (ssDNA) [45,46].As shown in Fig.4A,Jianget al.[47] reported a T7 exo-mediated signal amplification reaction for detection of thymine DNA glycosylase (TDG) enzyme activity.TDG is an enzyme in humans that selectively remove T from G/T mismatches.A hairpin structure DNA with 5′ overhangs and one G/T mismatch in the stem part is designed as the substrate for TDG recognition.In the presence of TDG, T base will be excised to abasic site in the stem part of hairpin substrate, and then the abasic site will be cleaved by endonuclease IV (Endo IV), resulting in the hairpin substrate converting to a dsDNA with a recessed 5′ end in the short sequence for T7 exo degradation, releasing the long sequence in the dsDNA.Subsequently, the long sequence will be hybridized with a fluorescence-quenched TaqMan reporter to form a new hybrid.T7 exo catalyzes a successive hydrolysis of the Taq-Man reporter in the hybrid, leading to the disappearance of FRET between the fluorescent dye and the quencher and the substantial activation of the fluorescence signal.The detection limit was estimated to be 0.00018 U/mL.This method exhibited selectivity,simple operation and excellent reproducibility.
Compared with exonuclease, endonuclease has a defined recognition site [48,49].Nb.BbvCI is an endonuclease can recognize an asymmetric sequence (5′-GCTGAGG-3′) [49].As shown in Fig.4B,Liet al.[50] proposed a Nb.BbvCI enzyme assisted-signal amplification strategy for visualized UDG activity assay.UDG can specifically recognize and hydrolyze the U bases in the stem of hairpin probe 1 (HP 1), leading to the instability of its stem, and resulting in the opening of HP1 to form a ssDNA which can hybridizes with hairpin probe 2 (HP 2) to form a DNA duplex containing a full recognition site for the Nb.BbvCI enzyme.After the specific cleavage of Nb.BbvCI, the released HP1 is able to hybridize with another HP 2 to induce the continuous cleavage of HP 2.As a result, the generated amount of G-riched quadruplex sequences bind with hemin to form a catalytically active G-quadruplex-hemin DNAzyme which can catalyze the H2O2-mediated oxidation of ABTS2−to the colored ABTS−, providing a visible signal for UDG activity detection.
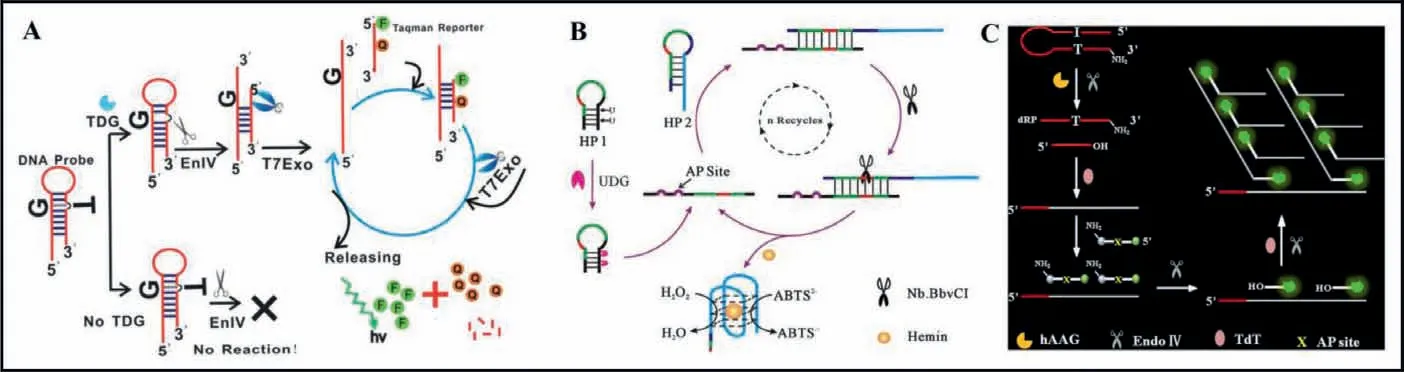
Fig.4.(A) Schematic illustration of T7 exonuclease-mediated amplification for TDG activity assay.Copied with permission [47].Copyright 2013, Royal Society of Chemistry.(B) Schematic illustration of Nb.BbvCI endonuclease assisted-signal amplification strategy for UDG activity assay.Copied with permission [50].Copyright 2014, Elsevier.(C)Schematic illustration of TdT polymerase assisted amplification method for hAAG activity assay.Copied with permission [52].Copyright 2019, Elsevier.
Terminal deoxynucleotidyl transferase (TdT) is a DNA polymerase which can catalyze the addition of deoxynucleotide triphosphates (dNTPs) to the 3′-OH terminus of a DNA sequence repetitively without the requirement of any DNA templates as primer [51].As shown in Fig.4C, Zhanget al.[52] established a TdT polymerase assisted amplification method for Human alkyladenine DNA glycosylase (hAAG) activity assay.The hairpin substrate for hAAG is modified with a deoxyinosine downstream of a 5′ terminus and a NH2in the 3′ termini to prevent TdT-activated nonspecific amplification.In the presence of hAAG, the damaged deoxyinosine base in the hairpin substrate can be recognized and cleaved to form an AP site which can be catalyzed by Endo IV to generate a free 3′-OH terminus.TdT is able to add multiple deoxyadenine triphosphate (dATPs) to the free 3′-OH termini sequentially to obtain the poly(A) sequences which can hybridize with signal probe with AP site to form a stable dsDNA.The AP site in ds-DNA may be catalyzed by Endo IV to generate a free 3′-OH termini which may initiate new TdT-mediated polymerization to form poly(A) sequences once again, and a distinct fluorescence may be produced.The poly(A) sequences can hybridize with new signal probe to initiate cycles of cleavage, extension, hybridization and release, eventually leading to an enhanced fluorescence signal.The assay method for DNA glycosylase is relatively simple and can be finished in a short time.
3.2.2.Strand displacement amplification (SDA) and exponential amplification reaction (EXPAR)
The amplification mechanism of SDA is based on the continuous nicking and polymerization/displacement process catalyzed by endonuclease and 5′−3′ exonuclease-deficient DNA polymerase in the presence of DNA template [53].Compared with SDA, the DNA template of EXPAR is composed of two identical nucleic acid sequences, and the products of EXPAR can be used as primers to induce new EXPAR for the generation of abundant triggers [54].
As shown in Fig.5, Zhanget al.[55] developed an actuating ligation-dependent SDA and EXPAR reaction for the detection of FPG activity.FPG could remove the damaged base 8-oxoG and cleave the hairpin substrate to form a hairpin cleavage product with 5′-PO4residue.Subsequently, the ligation probe hybridized with the hairpin cleavage product to form a new hairpin template in the presence of DNA ligase.The upper strand of the stem in hairpin template would initiate SDA reaction, producing the reporter probes.Moreover, the reporter probes could hybridize with the free ligation probe to initiate EXPAR reaction, eventually generating more reporter probes.The reporter probes produced by SDA and EXPAR hybridize with the signal probes to form stable dsDNA duplexes.And then the signal probes in the dsDNA duplexes could be digested by T7 exo to recover the FAM fluorescence.The digestion reaction of signal probes could proceed circularly to produce an increased fluorescence signal.
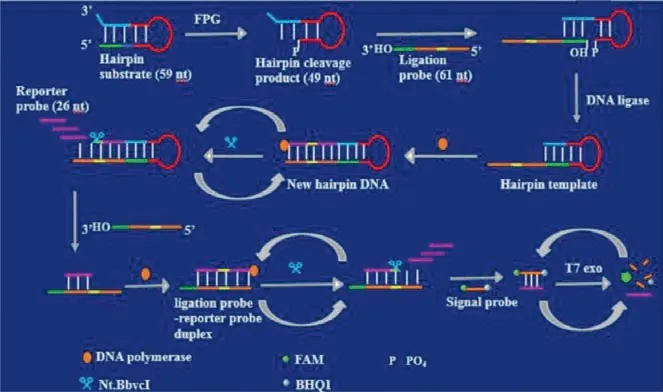
Fig.5.Schematic illustration of ligation-dependent SDA and EXPAR amplification method for FPG activity assay.Copied with permission [55].Copyright 2020, Elsevier.
3.2.3.Rolling circle amplification (RCA)
RCA is a simple and efficient isothermal enzymatic process that utilizes unique DNA and RNA polymerases to generate long ssDNA and RNA [56].RCA involves elongation of a DNA primer along with a circular DNA template by DNA polymerases, producing long ss-DNA with tens to hundreds of repetitive sequences that are complementary to the circular DNA template.Thus, the long ssDNA of RCA amplification products can be tailor-designed through manipulation of the circular DNA template.Multifunctional materials with diverse properties including biorecognition, sensing and imaging can easily be made by hybridizing RCA products with complementary oligonucleotides tethered with functional moieties including FAM, biotin, antibodies, and nanoparticles [57].RCA has been explored extensively to develop sensitive detection methods for DNA glycosylase activity [52,58-67].
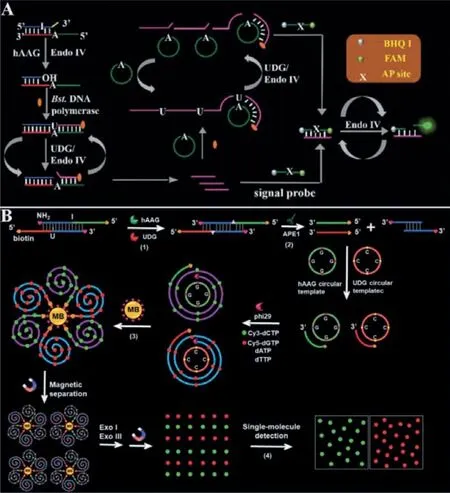
Fig.6.(A) Schematic illustration of PG-RCA for hAAG activity assay.Copied with permission [66].Copyright 2019, Royal Society of Chemistry.(B) Schematic illustration of RCA-driven encoding for simultaneous detection of hAAG and UDG activity.Copied with permission [67].Copyright 2020, Royal Society of Chemistry.
As shown in Fig.6A, Zhanget al.[66] developed a primer generation rolling circle amplification (PG-RCA) for hAAG activity assay.hAAG could specifically excise the mismatched deoxyinosine in hairpin substrate, releasing the damaged base to form an AP site.Subsequently, the AP site can be cleaved by Endo IV, resulting in the breaking of the hairpin substrate.And then, the cleaved hairpin substrate acts as a primer to initiate the SDA reaction in the presence of dNTPs including dATP, deoxyguanine triphosphates (dGTP),deoxycytosine triphosphates (dCTP) and dUTP, releasing primers.The primers hybridize with the circular template to initiate an exponential PG-RCA reaction, producing large amounts of primers.The primers resulting from the SDA and PG-RCA reaction can hybridize with the signal probes to form stable dsDNA, respectively.The signal probe in dsDNA may be cleaved by Endo IV, generating a distinct fluorescence signal and simultaneously releasing the primers.The released primers can hybridize with new signal probes circularly to generate an enhanced fluorescence signal.
The simultaneous detection of multiple DNA glycosylases will benefit the study of DNA damage repairing process and early clinical diagnosis, because some diseases exhibit multiple DNA glycosylation enzyme abnormalities.Some methods have been developed for simultaneous detection of multiple DNA glycosylases [67–71].As shown in Fig.6B, Zhanget al.[67] developed a RCA-driven encoding for simultaneous detection of multiple DNA repair enzymes based on the integration of single molecule detection for the first time.A bifunctional dsDNA substrate with one I base in one strand and one U base in the other strand was designed for hAAG and UDG recognition, respectively.In the presence of hAAG and UDG,they can specifically recognize I:T base pairs and U:A base pairs in the dsDNA substrate.The formed AP sites can be cleaved by apurinic/apyrimidinic endonuclease (APE1) to generate 5′-biotin labeled hAAG and by UDG primer to generate free 3′-OH terminus.The resultant two primers can hybridize with their respective circular templates to initiate RCA in the presence of phi29 polymerase and four kinds of deoxyribonucleotides (i.e., dATP, dTTP, Cy3-dCTP and Cy5-dGTP).Since the hAAG circular template contains only three types of bases (i.e., A, T, and G) and the UDG circular template contains only three types of bases (i.e., A, T and C), the hAAG amplification product includes only three types of bases (i.e., T,A, and C) and the UDG amplification product includes only three types of bases (i.e., T, A, and G).As a result, a large number of Cy3-modified dCTP are incorporated in the RCA product of hAAG and a large number of Cy5-modified dGTP in the RCA product of UDG according to base matching rule.After magnetic separation,the amplification products of hAAG and UDG with biotin at the 5′-termini are separated from the reaction solution and are subsequently digested into single nucleotides by exonucleases I and III.The Cy3 and Cy5 fluorescent molecules in the amplified products are released into the solution and subsequently quantified by total internal reflection fluorescence (TIRF)-based single-molecule detection for the quantification of hAAG and UDG, respectively.There is no spectral overlap between the emission of Cy3 and that of Cy5 because the maximum emission wavelength for Cy3 is 568 nm and that for Cy5 is 670 nm.
3.2.4.Loop-mediated isothermal amplification (LAMP)
In comparison with other nucleic acid amplification techniques,the sensitive loop-mediated isothermal amplification (LAMP) can achieve 109copies accumulated from less than 10 copies of input template under isothermal conditions within 1 h utilizing cyclic strand displacement amplification [72].LAMP employs a set of four or six primers to target distinct regions on double-stranded and only a single type of DNA polymerase.The reaction progress of LAMP contains a noncycling amplification step and a cycling amplification step.As shown in Fig.7, Zhanget al.[73] constructed a LAMP system for sensing of DNA glycosylases with zero background for the first time.A stem-loop DNA template with the damaged 8-oxoG and four linear primer probes (forward inner primers(FIP), forward outer primers (FOP), backward inner primers (BIP)and backward outer primers (BOP)) are designed in this strategy.The hOGG1 is a kind of bifunctional DNA glycosylase with both glycosylase and AP lyase activities, which can not only excise 8-oxoG in the DNA template to generate an AP site but also hydrolyze the phosphodiester bond at the AP site to generate a nucleotide gap, leading to opening the loop structure of the DNA template.In the presence of FIP, FOP and Bst.DNA polymerase, FIP will firstly hybridize with F2c in unfolded DNA template to initiate the strand displacement synthesis.And FOP (a few bases shorter and lower in concentration than FIP) can hybridize with FOPc in the unfolded DNA template simultaneously to initiate the strand displacement synthesis, producing a single-strand DNA which can form a new stem-loop structure through the hybridization between F1 and F1c.Similarly, BIP and BOP can hybridize with B2c and BOPc in the resultant one-stem-loop DNA, which can initiate the polymerization extension and successively release an ss-DNA that can form a double stem-loop structure (I) which contains F2c and B2 in the loops, respectively.After forming the double stem-loop DNAs, the self-primed polymerization extension at the 3′ end can be initiated to form a one-stem-loop DNA with only F2c in the loop which can hybridize with FIP and initiate the polymerization extension, producing a dsDNA intermediate with B2c in one loop.Subsequently, the dsDNA intermediate initiate the selfprimed polymerization extension, generating a one-stem-loop DNA(I) containing a loop with B2c and a double-stem-loop DNA (II)containing F2 and B2c in the loops.Subsequently, BIP can hybridize with the B2c in the loop to initiate polymerization extension, producing a dsDNA intermediate which contains one loop with F2c.And then, the dsDNA intermediate will initiate the polymerization extension to generate a one-stem-loop DNA containing a loop with B2c and a double-stem-loop DNA (I) containing F2c and B2 in the loops, respectively.Similarly, the self-primed polymerization extension can be performed repeatedly for the exponential amplification of DNA and the generation of large amounts of dsDNA products,which can be detected with SYBR Green I as the label-free fluorescence indicator for quantifying DNA glycosylase activity.The assay reaction is implemented in one tube with involvement of only a single type of polymerase under isothermal conditions, and the output fluorescence signal can be detected in a label-free manner,greatly reducing the assay costs.
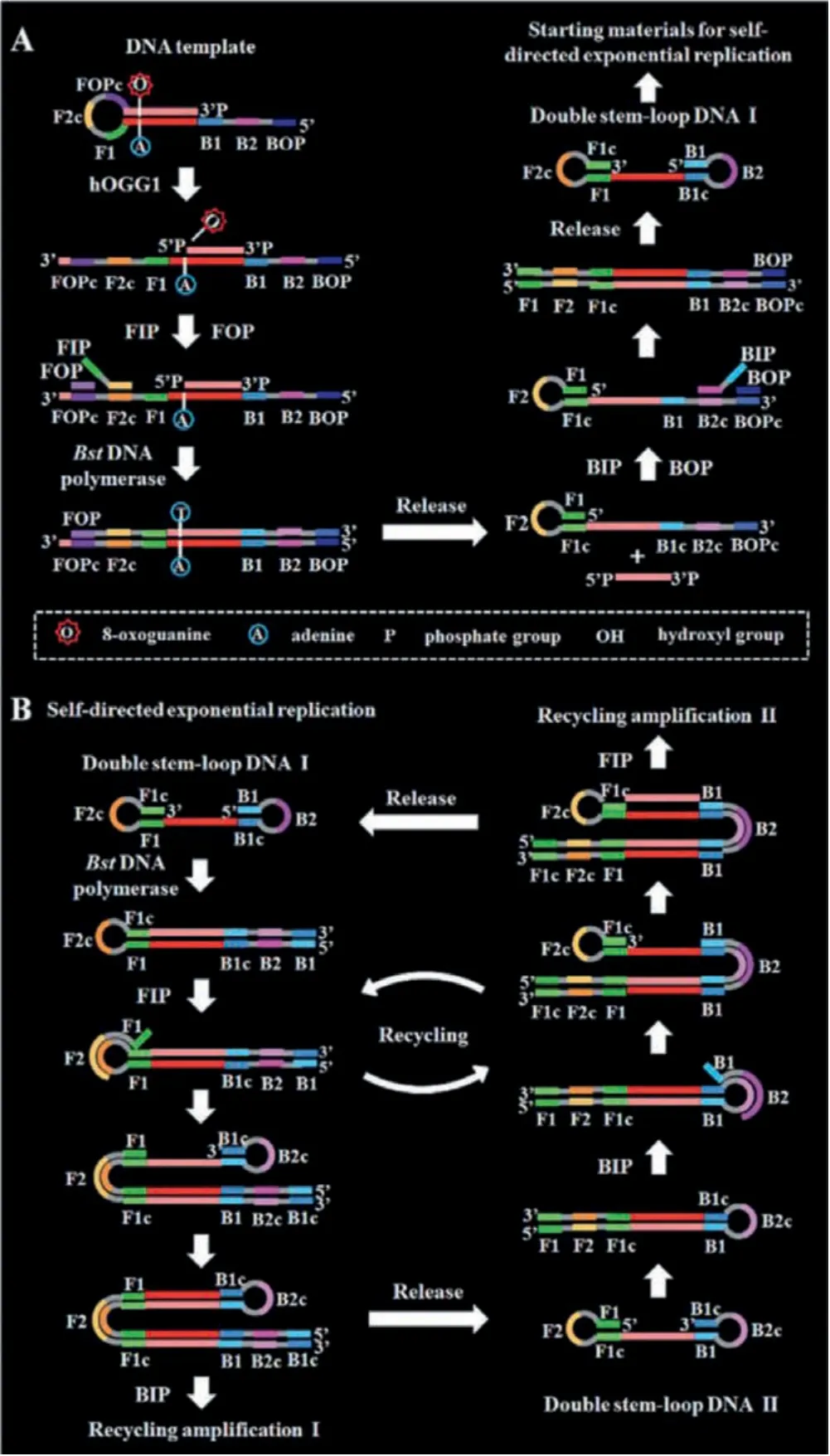
Fig.7.Schematic illustration of DNA glycosylase assay with LAMP.Copied with permission [73].Copyright 2020, Royal Society of Chemistry.
3.2.5.Enzyme-free strand displacement reactions (SDR)
Differences in sequence complementarity between multiple DNA strands can lead to the SDR.The thermodynamically driven SDR involves three elements: A substrate strand, an initiator strand, and an output strand.The substrate and output strands are specifically designed to partially hybridize to one another, leaving an exposed region on the substrate strand.This overhanging region is known as a toehold, which guides hybridization with the initiator strand.Upon introduction of the initiator strand, which is fully complementary to the substrate strand, the output strand is displaced from the substrate in a thermodynamically-driven migration process.This process results in the formation of a substrateinitiator duplex.In addition, the use of toeholds enables the construction of enzyme-free DNA reaction networks exhibiting complex dynamical behavior [74].In recent years, a variety of enzymefree strand displacement reactions have become a powerful tool for the detection of DNA glycosylase [75–84].
Hybridization chain reaction (HCR) is a simple and efficient isothermal amplification process proposed by Dirks and Pierce in 2004 [85].In a typical HCR, an initiator triggers a cascade of hybridization events between two species of DNA hairpins, leading to the formation of a nicked double helix with tens to hundreds of repeated units until the hairpins are exhausted.Given the advantages of its enzyme-free nature, efficient isothermal amplification,ultrahigh sensitivity and structural flexibility, HCR has emerged as a powerful molecular tool with versatile applications in biosensing, bioimaging, and biomedicine [86].As shown in Fig.8A, Fanet al.[75] reported a HCR method for UDG activity assay by coupling photoelectrochemical (PEC) and EC strategies.TiO2and Au nanoparticles were first modified sequentially on a clean indium–tin oxide (ITO) electrode to form an Au/TiO2matrix for immobilizing substrate DNA and blocking reagent of 6-mercapto1-hexanol(MCH).AgInS2quantum dots (AIS QDs) as PEC labels were bound covalently on the substrate DNA (sDNA), producing sensitization structure between the AIS QDs and the Au/TiO2matrix.In the presence of UDG, the U would be removed and produced an AP site which could be cleaved by Endo IV to release the PEC labels of AIS QDs from the electrode surface.In this case, the PEC signal of the probe electrode decreased because sensitization effect of the AIS QDs to the Au/TiO2matrix disappeared.After assistant DNA was then assembled on the electrode, H1 and H2 with ferrocene(Fc) molecules were introduced on the electrode via the HCR process, producing an evidently increased EC signal.Besides, as the long dsDNA produced by the HCR process, it has obvious steric hindrance to inhibit photogenerated electron transfer, the PEC signal further decreased.
Catalytic hairpin assembly (CHA), a programmable DNA circuit,was first proposed by Yinet al.in 2008 [87].A typical CHA reaction is initiated by single-stranded analytes to open the hairpin substrates, resulting in thermodynamically stable duplexes.Owing to its promising versatility, CHA is able to be applied for analysis of various biomarkersin vitroand living cells [88].As shown in Fig.8B, Jianget al.[76] developed a CHA strategy for sensitive detection of UDG activity.The ssDNA P2 containing both the U bases and trigger sequence was partly hybridized with the inhibition strand P1 to form P1-P2 dsDNA probe.In the presence of the UDG, U bases could be removed from P2, generating AP sites and leading to the liberation of P2′ from P1-P2, and the P2′ could initiate the signal amplification of CHA.The trigger strand P2′ could hybridize with the toehold in hairpin H1, resulting the opening of hairpin H1, exposing a new toehold which could hybridize with the toehold in hairpin H2.Since the H1-H2 complex was more stable than H1-P2′ hybridization, hairpin H2 could displace P2′ to form H1-H2 complex, and the single strand domains of H2 in H1-H2 complex were able to form G-quadruplex-N-methylmesoporphyrin IX (NMM) complex in the presence of monovalent ions for generating label-free fluorescence signal.As a result, the released P2′could trigger the next reaction cycle to consecutively generate H1-H2 complexes containing G-quadruplex structures, generating an enhanced fluorescence signal.
The DNA three way junctions (TWJs) are flexibly controlled DNA dynamic assemblies commonly consisting of three complementary oligonucleotide branches [89].Toehold-mediated strand displacement reaction (TSDR) with TWJs has the advantages of conformational stability and structural flexibility and is widely used for biosensing applications.As shown in Fig.8C, Xianget al.[77] developed a TSDR with TWJs approach for monitoring UDG activity.The DNA TWJ probes are immobilized on electrode surface via the thiol-Au bond.The two toeholds for the initiation of the TSDR are located in the DNA TWJ probe and the methylene blue-stranded 2/stranded 3 (MBS2/S3) duplex, respectively.The MB-S2 hybridized with stranded 1 (S1) to suppress TSDR, and protection probe (PP)is locked in the stem of the DNA TWJ.UDG can specifically recognized and hydrolyzed the U bases in the S1/MB-S2 duplex, the generated AP sites may lead to the significantly decreased melting temperature, resulting in releasing single stranded MB-S2.And then the released MB-S2 could bind with the first toehold region in the DNA TWJ probes and initiate the TSDR, resulting in the capture of MB-S2 on the surface of electrode and the liberation of PP and assistant probe (AP).The released PP further binds the second toehold of MB-S2/S3 duplex to trigger another TSDR to generate MB-S2.The released MB-S2 could circularly initiate the TSDRs, resulting in a significantly amplified current responses for achieving high sensitivity of UDG detection.
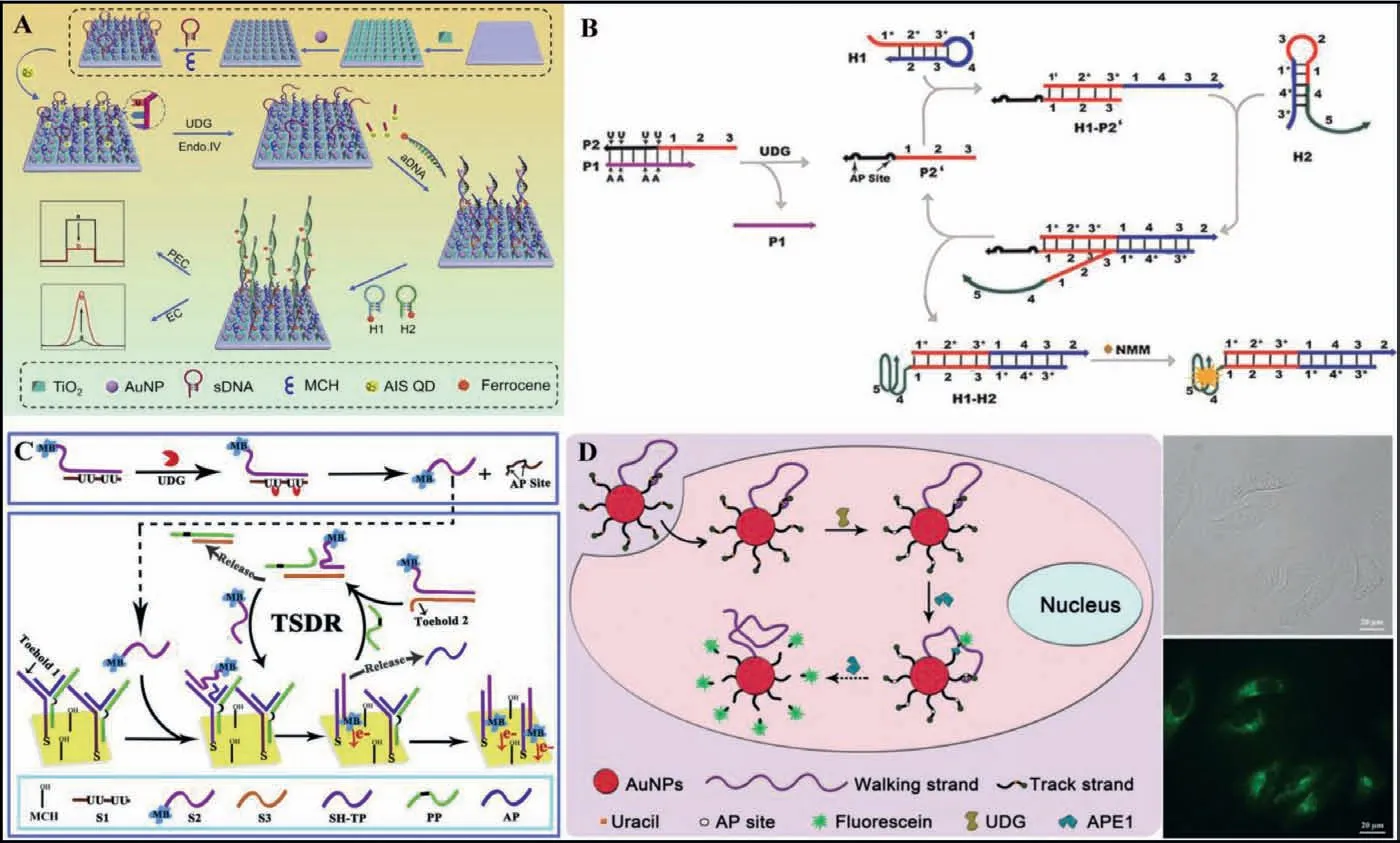
Fig.8.(A) Schematic illustration of HCR for UDG activity assay.Copied with permission [75].Copyright 2019, Elsevier.(B) Schematic illustration of CHA for sensitive detection of UDG activity.Copied with permission [76].Copyright 2015, Elsevier.(C) Schematic illustration of a TSDR with three way junctions approach for monitoring UDG activity.Copied with permission [77].Copyright 2018, Elsevier.(D) Schematic illustration of a DNA walker powered by endogenous enzymes for imaging UDG activity in living cells.Copied with permission [78].Copyright 2019, Royal Society of Chemistry.
DNA walkers are a unique class of dynamic DNA devices that move nucleic acid walkers progressively along designated one-,two-, or three-dimensional tracks.Because of the unique mechanical motion, dynamic interaction, and capabilities for signal amplification, programmable signal transduction, high directionality,and predictable analytical performance on the basis of Watson-Crick base paring rules, this class of dynamic DNA nanodevice has gained great attention from the analytical community in the recent years [90].As shown in Fig.8D, Jianget al.[78] reported a DNA walker powered by endogenous enzymes for imaging UDG activity in living cells.The DNA walker is constructed on a gold nanoparticle (AuNP) surface, constituted of walking strands and large amounts of fluorescein-labelled, U containing track strands.The fluorescence is quenched by AuNP.The walking strand is partially complementary to the track strand.The DNA walker and all its components can be readily taken up by living cells through nanoparticle-assisted endocytosis.Moreover, the nanoparticle surface-immobilized DNA can be protected from nuclease degradation.The U base in the track strand is removed under the action of endogenous UDG, generating an AP site which can be cleaved under the action of endogenous APE1.As a result, the fluorescein is released and the fluorescence is restored.Then, the walking strand continues hybridizing with the next track strand and initiates the next cycle of track strand cleavage and fluorescein release, moving along the three dimensional (3D) surface until the entire track strands are consumed.Due to the resistance to nonspecific nuclease degradation and unique amplification ability of the walkers, this method can be used to detect the UDG activity in living cells.However, the sensitivity is limited because of the unprotected DNA probe and the amplification inside the cells is difficult.
3.3.Nanomaterial-based signal amplification
With the development of material science and nanotechnology,a series of new nanomaterials has been successfully designed and synthesized [91–94].Due to their unique sizes and shapes, nanomaterials possess some attractive properties such as optical emitters and carrier platforms.Moreover, nanomaterials can be easily biofunctionalized, making them powerful building materials for the construction of efficient signal amplification biosensors [95].Nanomaterial-based biosensors have been widely applied for the sensitive and selective detection of DNA glycosylase activity [35,96-100].
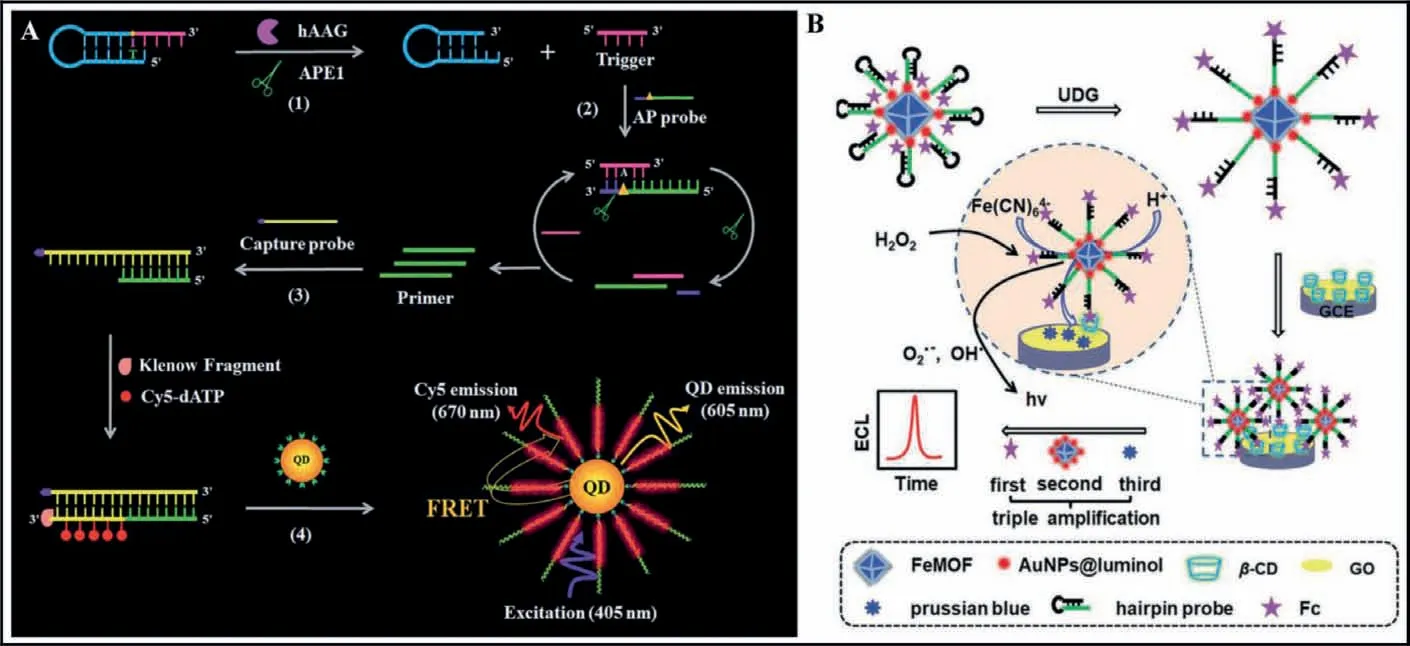
Fig.9.(A) Schematic illustration of a single QD-based nanosensor for detection of hAAG.Copied with permission [99].Copyright 2019, Royal Society of Chemistry.(B)Schematic illustration of a MOF-based signal amplification for the detection of UDG.Copied with permission [100].Copyright 2020, Royal Society of Chemistry.
Semiconductor quantum dots (QDs) exhibit unique optical and physical properties (e.g., high brightness, high quantum yield, good stability against photobleaching, narrow emission bands and sizetunable emission spectra) that are not shared by organic dyes and fluorescent proteins, and their dimensions are comparable to those of biomolecules [101–103].As shown in Fig.9A, Zhanget al.[99] developed a single QD-based nanosensor for ultrasensitive detection of hAAG using FRET from the QD to Cy5.In the presence of the hAAG, it can specifically recognize the I base and cleaves theN-glycosidic bond between the sugar and the I base, releasing the I base to form an AP site which can be cleaved by APE1,generating a trigger and a stable stem-loop DNA fragment.The resultant triggers can hybridize with the AP probes to form the ds-DNAs which can be cleaved circularly by APE1, producing a large number of primers with 3′-OH.The primers hybridize with capture probe to form a partially complementary dsDNA.And then the primers extend along with the capture probe stable dsDNAs with the incorporation of multiple Cy5 molecules in the presence of the Klenow fragment, Cy5-dATP, dCTP, dGTP and dTTP.As a result, the QD-dsDNA-Cy5 nanostructure can be formedviaspecific biotin–streptavidin binding, and efficient FRET occurs with the QD as the donor and Cy5 as the acceptor under the excitation of 405 nm, releasing the fluorescence signal.
Metal-organic frameworks (MOFs) are composed of metal clusters/ions which are connected by special organic linkers, exhibiting superior physical and chemical properties [104].Due to their unique properties including large surface areas, large pore volumes,tunable surface chemistry and pore size, and multiple topologies,MOFs have been used as promising porous materials with wide applications in adsorption and separation, sensing, catalysis, drug delivery and imaging [105].As shown in Fig.9B, Zhanget al.[100] demonstrated a MOFs nanomaterial-based signal amplification for the detection of UDG by host–guest recognition.The hairpin probe is modified with Fc in the 3′ end as the first signal amplification redox and a thiol group in the 5′ end for immobilization on the FeMOF/AuNPs@luminol as the second signal amplification platform.In the presence of UDG, six U bases in the stem of the hairpin probe can be removed from the hairpin DNA stem to produce six AP sites, leading to an ssDNA with Fc in the 3′ end.Theβ-CD on the electrode can recognize Fcviahost-guest interaction.Subsequently, Fc (Fc+) catalyzes the reaction of luminol radicals with hydroxyl radical to form 3-aminophthalate, resulting an enhanced ECL signal.Moreover, the addition of acid and K4Fe(CN)6enables their reaction with Fe3+in a FeMOF, resulting in the production of prussian blue which can catalyze the formation of 3-aminophthalate in the presence of H2O2in alkaline solution, enabling the ECL signal amplification.
4.Conclusion and prospects
Accurate quantification of DNA glycosylase activity can be applied for the measurement of enzyme kinetic parameters and the screening of inhibitors for DNA glycosylase, playing a crucial role in glycosylase-related function study and disease prognosis.In this minireview, we summarize the recent advances in DNA glycosylase assays including amplification-free assays and amplificationassisted assays (Table 1).The amplification-free assays are much simple and enable rapid detection of DNA glycosylases.Compared with amplification-free assays, amplification-assisted assays (PCR and isothermal nucleic acid amplification approaches) possess the advantages of high sensitivity down to single cells for the detection of cellular DNA glycosylases activity.Importantly, two DNA glycosylases can be detected simultaneously by rationally designing appropriate DNA probes.The development of DNA glycosylase assay with the advantages of rapidity, simple operation, low cost,high sensitivity and selectivity, and high throughput at the same time remains a great challenge.Great efforts should be put into improving the detection sensitivity for the low abundance of DNA glycosylase in single cell.It is urgent to develop novel signal amplification method with spontaneous reaction for DNA glycosylase assays without the involvement of protease in complex cell environment.In addition, one particular cellular function may rely on the cooperation of a variety of different DNA glycosylase, and the simultaneous detection of more than two DNA glycosylases is highly desired, which can be achieved by functionalizing various specifically probes towards different DNA glycosylase.With the development of separation methods based on capillary electrophoresis, chromatography and the discovery of novel nanomaterials with efficient labeling strategies, we envision that the development of high-throughput assays of DNA glycosylases may greatly facilitate clinical diagnosis and drug screening in the near future.
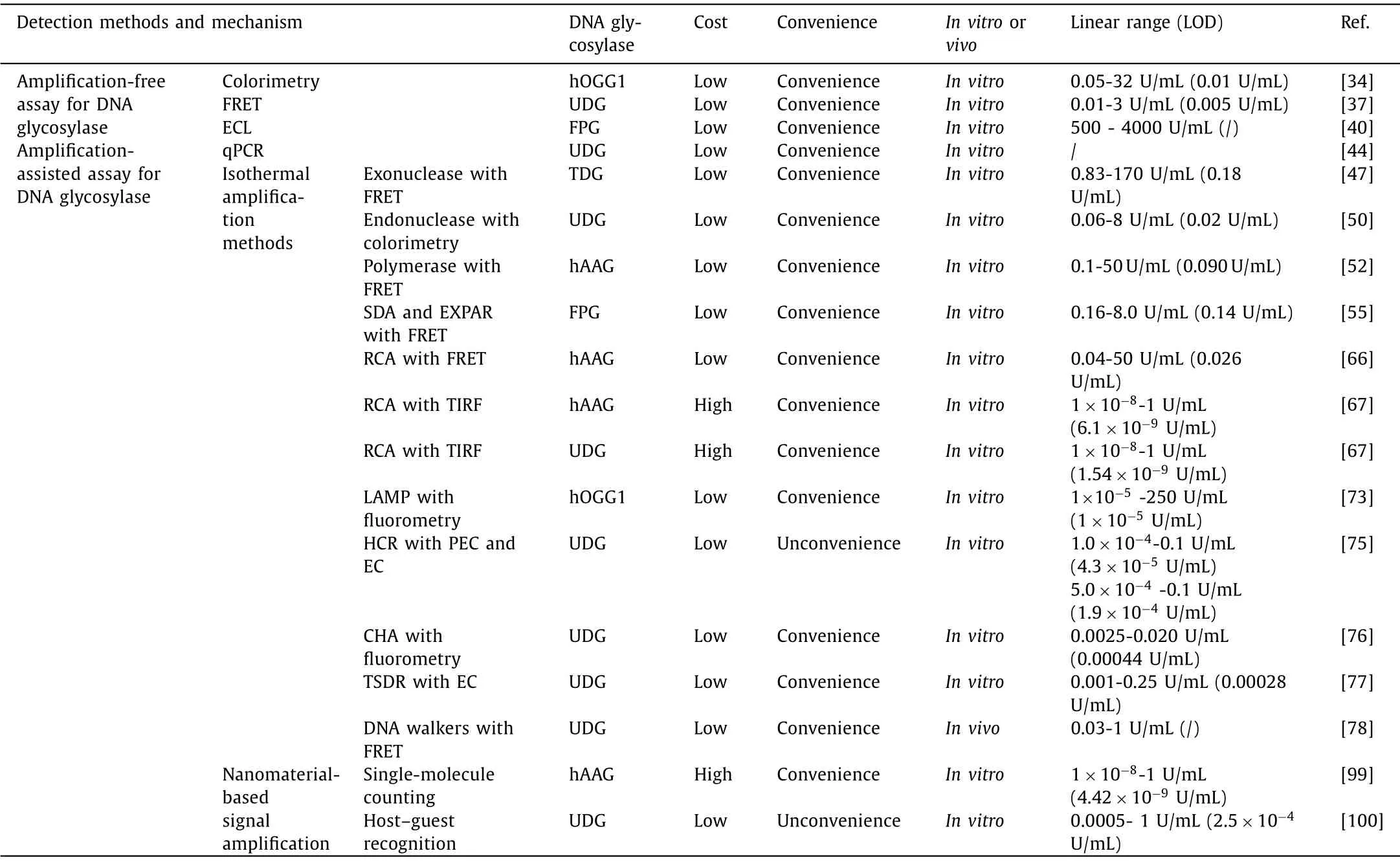
Table 1.Recent advances in DNA glycosylase assays.
Declaration of competing interest
The authors declare no competing financial interest.
Acknowledgments
The authors are grateful for the financial support from the National Natural Science Foundation of China (No.21874060) and the Fundamental Research Funds for the Central Universities (No.lzujbky-2021-it15).
 Chinese Chemical Letters2022年8期
Chinese Chemical Letters2022年8期
- Chinese Chemical Letters的其它文章
- Adsorptive removal of PPCPs from aqueous solution using carbon-based composites: A review
- A review on hollow fiber membrane module towards high separation efficiency: Process modeling in fouling perspective
- Chiral pillar[n]arenes: Conformation inversion, material preparation and applications
- Recent progress in carbon-based materials boosting electrochemical water splitting
- Working principle and application of photocatalytic optical fibers for the degradation and conversion of gaseous pollutants
- Crystal facet-dependent electrocatalytic performance of metallic Cu in CO2 reduction reactions