
Selective Ni-catalyzed cross-electrophile coupling of alkynes,fluoroalkyl halides, and vinyl halides
2022-09-16 05:25YubeiDaiFangWangShengqingZhuLinglingChu
Chinese Chemical Letters 2022年8期
Yubei Dai, Fang Wang, Shengqing Zhu, Lingling Chu
State Key Laboratory for Modification of Chemical Fibers and Polymer Materials, Center for Advanced Low-Dimension Materials, College of Chemistry,Chemical Engineering and Biotechnology, Donghua University, Shanghai 201620, China
ABSTRACT We report a Ni-catalyzed three-component cross-electrophile coupling of alkynes with alkenyl halides and fluoroalkyl halides to generate fluoroalkyl-incorporated 1,3-dienes.This mild and operationally simple protocol is distinguished by its broad substrate scope and excellent chemo-, regio-, and stereo-selectivity,offering a new and organometallic agent-free platform for the construction of fluoroalkyl-incorporated diene motifs.Preliminary mechanistic studies have been conducted to probe the potential reaction pathway.
Keywords:Fluoroalkylation Cross-electrophile coupling Nickel catalysis Difunctionalization Alkynes
Due to the unique properties of the fluorine atom, the selective incorporation of fluorine and fluoroalkyl groups into organic molecules has attracted significant attention [1–3].Impressive progress has been made in the direct fluoroalkylation of aromatic motifs during the last decade [4–9], however, methodologies for the straightforward construction of fluoroalkyl-incorporated alkenes, particularly multisubstituted alkenes and dienes, remained underexplored [10–13].Currently known protocols mainly focus on two-component systems involving fluoroalkylative cross-couplings between alkenyl nucleophiles (or electrophiles) and fluoroalkylating agents [14–21].Recently, increasing attention has been drawn to transition metal-catalyzed three-component fluoroalkylative functionalization of alkynes [22–34], that allows for the installation of fluoroalkylated alkenes with a simultaneous introduction of another C−C bond (Fig.1A).Such a three-component strategy not only offers a new retrosynthetic route for the assembly of fluoroalkylated alkenes, but also enables a rapid increase in the molecular complexity of these valuable motifs.However, most of these reactions focus on the couplings with aryl, alkynyl, or cyanide nucleophiles in the presence of palladium or copper complexes.However, the similar strategy has not been applied to the construction of fluoroalkylated dienes with vinyl parters, particularly enabled by non-noble transition metal catalysts.
Transition metal-catalyzed dicarbofunctionalization of alkynes has been identified as a powerful platform for the construction of multisubstituted alkenes, where two different carbon-centered fragments have been appended into alkynes in one single operation [35–41].Generally, these transformations have been accomplished by selectively coupling alkynes with an electrophile and a nucleophile that typically is an organometallic agent.Recently, nickel-catalyzed cross-electrophile couplings with two electrophiles in the presence of stoichiometric reductants, that avoid the use of sensitive organometallic agents, have emerged as attractive strategy for 1,2-difunctionalization of unsaturatedπ-bonds[42–46].Nevertheless, the majority of these processes focus on the utilization of olefins to forge C(sp3)-alkyl or C(sp3)-aryl bonds[40,41,46–50].In contrast, similar transformations of alkynes remain underexploited, probably due to the problematic step of radical addition to alkynes [51] as well as the inherent chemoselectivity challenge.To date, only two examples of Ni-catalyzed threecomponent reductive dicarbofunctionalization of alkynes with two electrophiles have been disclosed [52,53].To the best of our knowledge, no examples of catalytic carbo-alkenylation of alkynes with alkenyl halides to forge 1,3-dienes, that are prevalently found in many biologically active natural products and pharmaceuticals as well as serving as versatile synthetic intermediates for drugs,dyes, and functionalized polymers [54–62], have been disclosed.As part of our continuing interest in Ni-catalyzed difunctionalization of unsaturatedπ-bonds [63–73], we herein report a new and efficient three-component fluoroalkyl-alkenylation of alkynes with alkenyl halides and fluoroalkyl halidesvianickel-catalyzed cross-electrophile coupling (Fig.1B).This protocol enables straightforward access to stereodefined fluoroalkyl-incorporated 1,3-dienes from readily available starting materials under mild conditions.
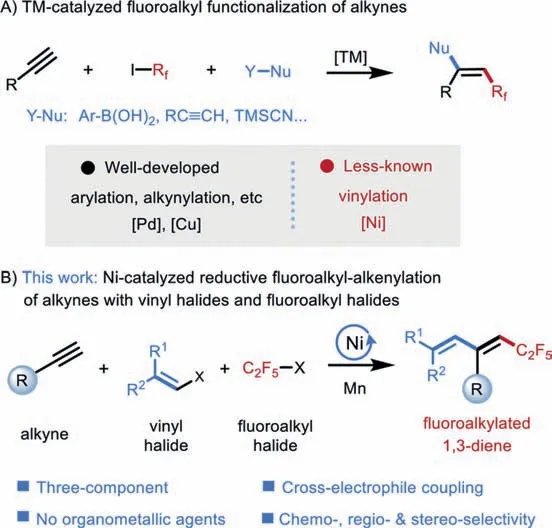
Fig.1.Ni-catalyzed cross-electrophile fluoroalkyl-alkenylation of alkynes.
Evaluation of this Ni-catalyzed reductive fluoroalkylalkenylation strategy was examined with 2-methyl phenylacetylene 1, benzyl (E)-3-iodoacrylate 2, and C2F5I 3 as model substrates(Table 1).In the presence of catalytic NiCl2·DME and 4,4′-di-tertbutyl-2,2′-bipyridine (dtbbpy) with Mn as reductant and TMSCl as additive, we were pleased to find that the three-component reaction of 1 with 2 and C2F5I ([1∼1.2 mol/L] in diglyme) underwent smoothly to afford (E)-C2F5-diene product 4 in 92%yield (entry 1).Both Ni(II) and Ni(0) catalysts were effective for this transformation, whilst simple NiCl2·DME proved to be optimal (entries 1−4).Solvents also play an important effect to reaction efficiency.THF turned out to be the best solvent,while running the reaction in other polar or nonpolar solvents resulted in decreased and even sluggish efficiency (entries 5−8).Switching to other commonly employed reductants such as Zn and TDAE (tetrakis(dimethylamino)ethylene) led to no formation of the desired products (entry 11).Control experiments further confirmed that nickel catalyst, ligand, and reductant were all required for the desired transformation, as no detection of desired products in the absence of each of them (entry 12).The addition of TMSCl as an additive, which might facilitate the activation of Mn [74–76], was found to be beneficial to the reaction efficiency(entry 13).Excellent regio- and stereo-selectivity is observed,and neither (Z)-isomers nor regioisomers were detected in all cases.
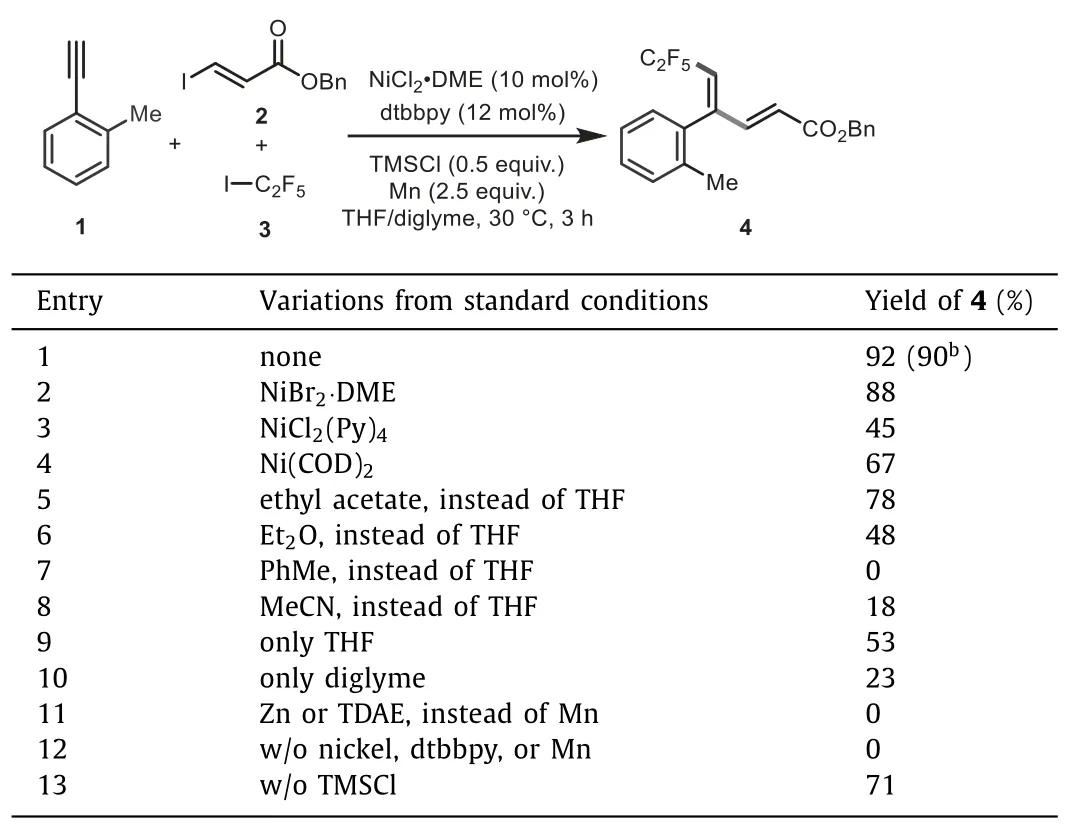
Table 1 Reaction optimizations.a
With the optimal reaction conditions in hand, we began to explore the generality of this Ni-catalyzed three-component fluoroalkyl-alkenylation reaction with respect to various alkynes.As shown in Scheme 1, a wide range of terminal arylalkynes bearing electron-withdrawing, -donating, or -neutral substituents on the aromatic rings all underwent efficient cross-couplings with alkenyl iodide 2 and C2F5I, furnishing the desired 1,3-diene product with moderate to high yields and excellent chemo-, regioand stereoselectivity (4−20, 65%−90% yields).The mild conditions tolerate many functional groups, including ethers, trifluoromethylates, cyanos, and halides.Generally,ortho-substituted arylalkynes performed with slightly higher efficiency thanpara-ormeta-substituted ones.We reasoned that the steric hindrance ofortho-substituents might help to slow down the undesired alkyne trimerization process [77].Heteroaryl-incorporated alkynes, exemplified by thiophenes, functioned as efficient coupling partners to afford the corresponding 1,3-dienes in moderate yields (21 and 22,45% and 67% yields, respectively).Notably, internal alkynes such as prop-1-yn-1-ylbenzene also worked well in this protocol, yielding diene 23 with excellent regio- and stereoselectivity.However,aliphatic alkynes were incompetent substrates in this Ni-catalyzed reductive protocol.
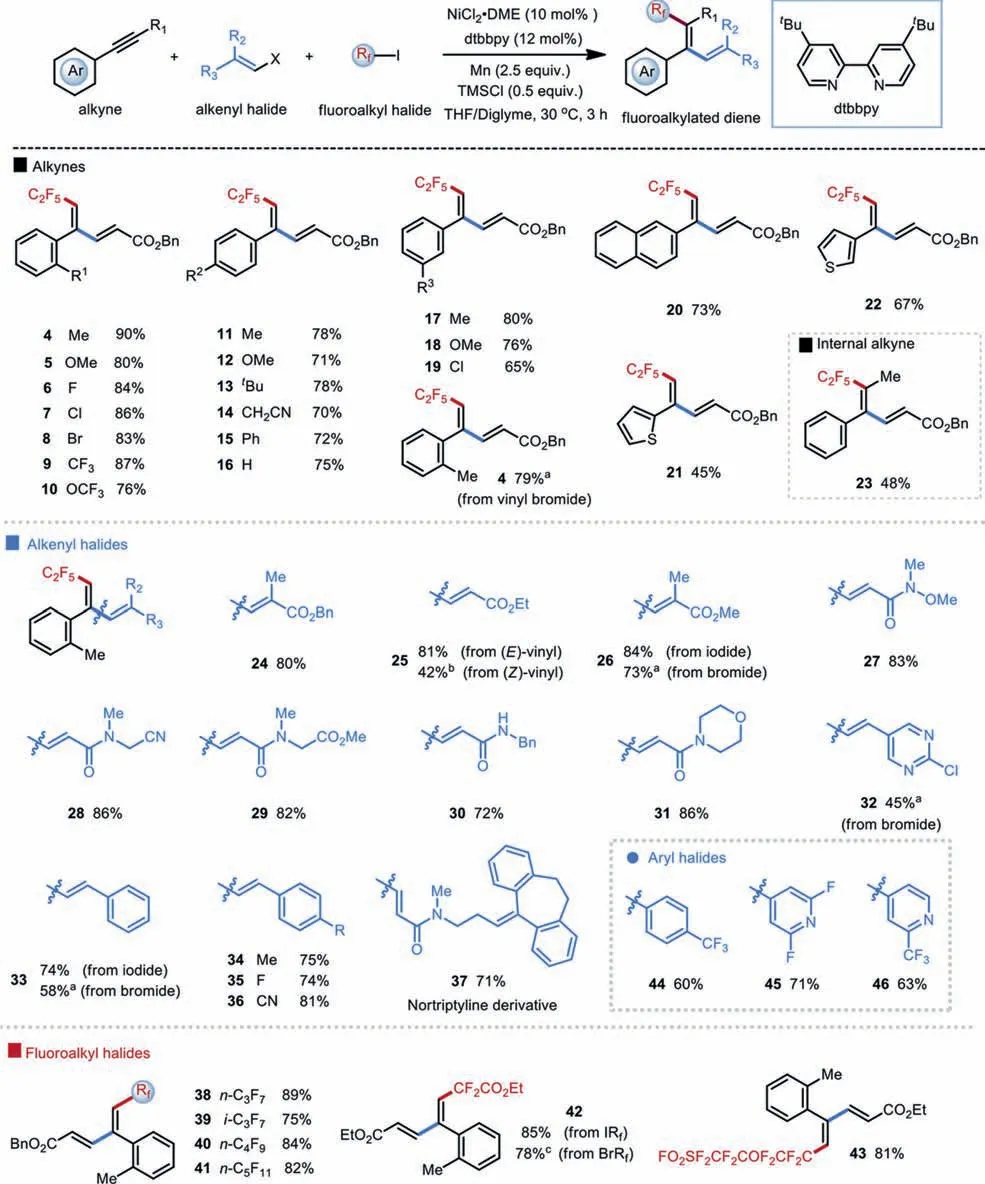
Scheme 1.Substrate scope of Ni-catalyzed three-component reductive fluoroalkyl-vinylation of alkynes.Reaction conditions: alkyne (0.2 mmol), alkenyl iodide (0.1 mmol),fluoroalkyl iodide (0.2 mmol), NiCl2·DME (10 mol%), dtbbpy (12 mol%), Mn (0.25 mmol), TMSCl (0.05 mmol), THF/diglyme = 1:1 [0.17 mol/L], 30 °C, 3 h.Isolated yields.a With alkenyl bromide. b With ethyl (Z)−3-iodoacrylate. c With ethyl bromodifluoroacetate.
Next, we evaluated the scope of the alkenyl halide component in this protocol.A series of substitutedβ-iodo acrylates were applicable with good efficiency (24−26, 42%−84% yields).Installation of substitutions on theα-position ofβ-iodo acrylates has no dramatic effect on the reaction efficiency (24 and 26, 80% and 84% yields, respectively).Interestingly, both (E)- and (Z)-alkenyl iodides worked in this protocol and yielded the same (E)-C2F5-dienes with excellent selectivity, whilst (Z)-alkenyl iodides showed much lower efficiency compared to theirtrans-isomers (25, 81%vs.42%).Furthermore,β-iodo acrylamides with various substituents were also suitable partners, furnishing the desiredδ-C2F5conjugated amides under mild conditions (27−31, 72%−86% yields).Notably,β-iodo acrylamides derived from nortriptyline, an anti-depressant drug [78], can be selectively coupled with alkyne 1 and C2F5I with good efficiency (37, 71% yield).Pleasingly, this reductive protocol could be further expanded toβ-iodo/bromo aryl- and heteroaryl alkenes with moderate to good efficiency (32−36, 45%−81% yields).Besides vinyl iodides, vinyl bromides also functioned as efficient coupling partners, albeit with slightly decreased yields (4, 79%vs.90%; 26, 73%vs.84%; 33, 58%vs.74%).Pleasingly, heteroaryl and aryl halides also proved to be competent coupling partners in this Ni-catalyzed reductive coupling protocol, delivering trisubstituted fluoroalkylated alkenes with moderate yields and excellent transselectivity (44−46, 60%−71% yields).
Finally, we investigated the scope of perfluoroalkyl iodides under the optimal conditions.A variety of fluoroalkyl iodides can serve as the competent coupling partners, affording the fluoroalkylated 1,3-dienes with moderate to high efficiency and excellent selectivity (38−41, 75%−89% yields).Nevertheless, fluoroalkyl iodides with the longer carbon chain demonstrated slightly decreased yields, probably due to their poorer solubility.Reaction with FSO2CF2CF2OCF2CF2I proceeds with high efficiency, leaving the SO2F group untouched (43, 81% yield).Moreover, both iodoand bromo–difluoroiodoacetate were applicable to couple with alkyne 1 and alkenyl iodide 2 with good efficiency (42, 85% and 78% yield, respectively).
To further demonstrate the usefulness of our catalytic radical domino protocol, we carried 1,3-diene compounds for a diverse of synthetic derivations, as shown in Scheme 2.Selective reduction of compound 33 with H2in the presence of catalytic Pd/C delivered trisubstituted alkene 47 in 70% yield (condition a) [79].Product 4 was easily converted into corresponding alkenyl acid 48 [69].amide 49 [80], and allyllic alcohol 50 [81]viaclassical hydrolysis or nucleophilic additions b−d.Furthermore, selective reduction of the ester group of 4 with DIBAL-H afforded dienyl allyl alcohol 51 in 78% yield (condition e) [82].[4 + 2] Cycloaddition of 51 with triazole dione (PTAD) gave C2F5-incorporated pyridazine dione 52 in 82% yield (condition f) [83].Reaction of 51 with vinylmagnesium bromide in the presence ofnBuLi underwent a sequential nucleophilic addition/defluorination process to deliver fluorinated diene 53 in 85% yield (condition g) [84,85].
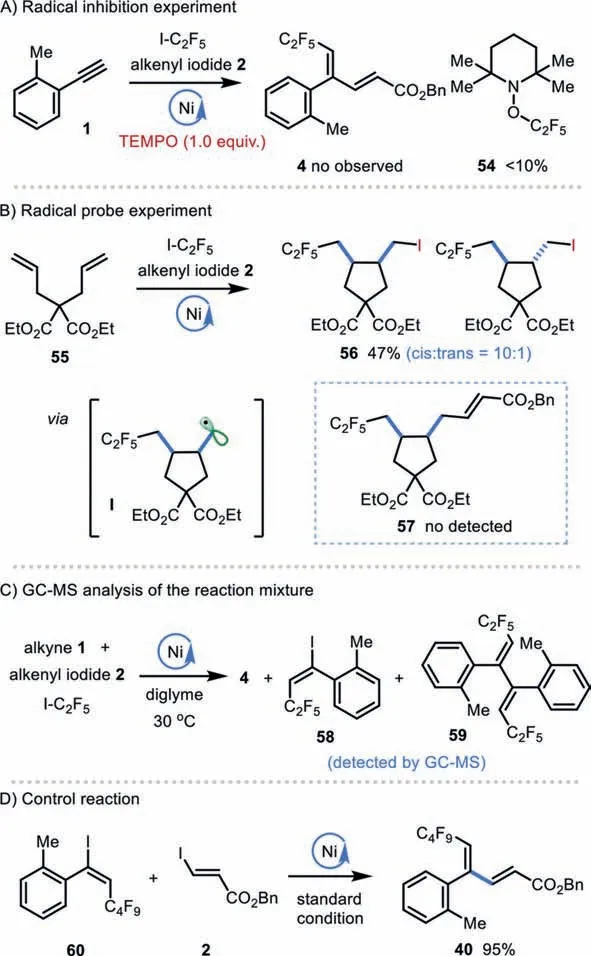
To shed some light on the potential reaction pathway of this novel catalytic reductive fluoroalkyl-alkenylation reaction, we have conducted several preliminary mechanistic experiments.The addition of 1 equiv.of TEMPO, a commonly employed radical inhibitor,into the template reaction system completely shut down the desired cross-coupling reaction, with only detection of TEMPO−C2F5adduct 54 (Scheme 3A) [86].Radical probe reaction with 1,5-diene was next explored (Scheme 3B).While the reaction of 55 with alkenyl iodide 2 and C2F5I under the optimal conditions gave the cyclized alkyl iodide 56 in 47% yield, with no observation of the desired alkyl-alkenylation product 57.We assume that 56 could be generated via an iodide transfer of alkyl radical I, which is formedviaa radical addition followed by 5-exo radical cyclization.Thecis/transratio of 56 (cis/trans= 10:1) also matches the involvement of radical intermediates, where thecisselectivity could attribute to the stabilization of the conformation for the cyclization transition state (Scheme 3B) [69,87].Interestingly, GC–MS analysis of the reaction mixture (in diglyme) detected the formation of alkenyl iodide 58 and a trace amount of dimer 59, further supporting the involvement of vinyl radical species (Scheme 3C).Moreover, time course studies of this reaction revealed the product formation was accompanied by generation of alkenyl iodide at the early stage,and alkenyl iodide was gradually converted into the final product at the late stage (see Supporting information).To further determine whether vinyl iodide could be a reactive intermediate, we subjected pre-prepared (E)-C4F9-alkenyl iodide 60 into the reaction system.The reaction of 60 with alkenyl iodide 2 in the presence of Ni(II)/dtbbpy/Mn furnished 1,3-diene 40 in 95% yield, suggesting that alkenyl iodide could be a reactive intermediate in this catalytic reductive system (Scheme 3D).
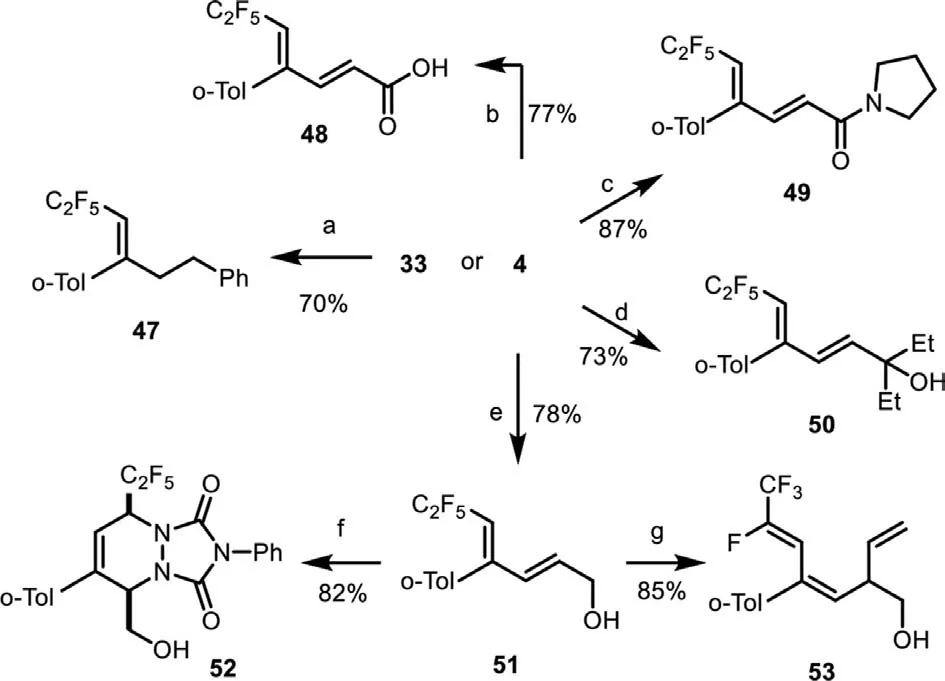
Scheme 2.Synthetic applicability.(a) Pd/C (10 mol%), H2 (balloon), MeOH, r.t.,12 h;(b) K2CO3, MeOH, r.t., 12 h; (c) LiClO4, pyrrolidine, r.t., 1 h; (d) EtMgBr, THF, −78 °C to r.t.; (e) DIBAL-H, CH2Cl2, −78 °C to r.t.; (f) 4-phenyl-3H-1,2,4-triazole-3,5(4H)–dione (PTAD), 1,2-dichloroethane, r.t.; (g) nBuLi, vinylmagnesium bromide, n-hexane,−78 °C to 90 °C.
Scheme 3.Mechanistic studies.
Scheme 4.Proposed reaction pathway.
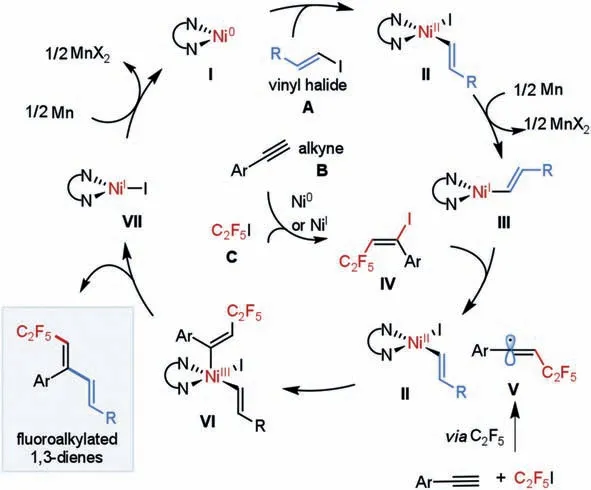
Based on these experimental results as well as previous literature [48,88–93], we proposed a reaction pathway for this Nicatalyzed reductive fluoroalkyl-alkenylation reaction, as depicted in Scheme 4.Initially, precatalyst Ni(II) is reduced by Mn to afford the active Ni(0) species I.Oxidative addition of vinyl halide A to Ni(0)gives (vinyl)Ni(II)-X II, which is then single-electron reduced by Mn to yield (vinyl)Ni(I) intermediate III.At the same time, alkyne B undergoes atom transfer radical addition (ATRA) with fluoroalkyl iodide C, assisted by Ni(0) or Ni(I) species, to furnishE-alkenyl iodide IV.At this juncture, we surmise that a SET event between(vinyl)Ni(I) and alkenyl iodide IV affords (vinyl)Ni(II) II and alkenyl radical V.Alternatively, direct radical addition of fluoroalkyl radical to alkyne also produces alkenyl radical V.V then combines with Ni(II) II generates Ni(III) species VI, which undergoes facile reductive elimination to furnish the desired fluoroalkylated 1,3-diene product as well as Ni(I) VII.Finally, SET reduction of Ni(I) VII in the presence of Mn would regenerate Ni(0) to close the catalytic cycle.In this reaction, the regioselective outcome is mainly steered by the addition of fluoroalkyl radicals to alkynes; while the excellenttrans-stereoselectivity could be attributed to the rapid inversion ofE/Zalkenyl radical V and a faster combination of Ni(II) II with less sterically hinderedE-alkenyl radical [94,95].
In conclusion, we have reported an efficient and selective threecomponent cross-electrophile fluoroalkyl-alkenylation of alkynes with fluoroalkyl halides and alkenyl halidesvianickel catalysis.This mild protocol enables the simultaneous incorporation of fluoroalkyl and alkenyl units, providing the straightforward approach to fluoroalkylated 1,3-dienes from readily available starting materials with excellent chemo-, regio- and stereo-selectivity.The reaction works well with a broad range of terminal and internal arylalkynes, alkenyl halides, and fluoroalkyl halides.Mechanistic studies by radical probes and time course studies indicate that this reaction could proceedviaa Ni(0)/Ni(I)/Ni(II)/Ni(III) cycle.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors are grateful for financial support provided by the National Natural Science Foundation of China (Nos.21991123,21971036, 21901036) and the Shanghai Rising-Star Program (No.20QA1400200).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.12.050.
 Chinese Chemical Letters2022年8期
Chinese Chemical Letters2022年8期
- Chinese Chemical Letters的其它文章
- Adsorptive removal of PPCPs from aqueous solution using carbon-based composites: A review
- A review on hollow fiber membrane module towards high separation efficiency: Process modeling in fouling perspective
- Recent advances in DNA glycosylase assays
- Chiral pillar[n]arenes: Conformation inversion, material preparation and applications
- Recent progress in carbon-based materials boosting electrochemical water splitting
- Working principle and application of photocatalytic optical fibers for the degradation and conversion of gaseous pollutants