
Electro-reductive C-H cyanoalkylation of quinoxalin-2(1H)-ones
2022-09-16 05:25LingDingKaikaiNiuYuxiuLiuQingminWang
Chinese Chemical Letters 2022年8期
Ling Ding, Kaikai Niu, Yuxiu Liu, Qingmin Wang
State Key Laboratory of Elemento-Organic Chemistry, Research Institute of Elemento-Organic Chemistry, College of Chemistry, Frontiers Science Center for New Organic Matter, Nankai University, Tianjin 300071, China
ABSTRACT Herein, we report a practical electro-reductive protocol for the direct C–H cyanoalkylation of quinoxalin-2(1H)-ones via iminyl radical-mediated ring opening.These mild reactions proceed under metal-,reductant-, and reagent-free conditions to provide synthetically useful cyanoalkylated quinoxalin-2(1H)-ones.
Keywords:Electro-reductive C-H cyanoalkylation Reductant-free Quinoxalin-2(1H)-ones Ring-opening
The cyanoalkyl moiety is of great significance in organic synthesis, it represents one privileged class of structural scaffolds in several nitrile-containing pharmaceuticals (Fig.1) [1,2].Therefore,efforts to introduce a cyanoalkyl group into structurally diverse molecules have attracted much attention [3,4].
Recently, the ring-opening reaction of cyclobutanone oxime derivatives, which has emerged as a powerful tool to realize cyanoalkylation reactions, comes into the spotlight [5–9].For instance, Nishimura and Uemura [10,11] developed a palladiumcatalyzed system using the ring strain of the cyclobutane skeleton to deliver nitriles.In addition, the pioneering work conducted by Boivinet al.[12,13] utilized cyclobutanone sulfenylimines and carboxymethyl oximes to trigger C-C bond cleavage with radical initiators or UV irradiation.Other investigations have also involved transition metal [14–20] or photoredox catalysis (Scheme 1A) [21–34].Nevertheless, the addition of sophisticated metal catalysts and precious metal-based catalysts in photocatalytic reactions limits their practical applications.
Electrochemical technique has long been hailed as an environmentally benign method due to their inherent ability to achieve redox reactions sustainably [35–38].To date, the direct electroreduction of cyclobutanone oxime derivatives has not been achieved.Therefore, building upon our previous research in organic radical chemistry [39–42], we now report a mild electrochemical protocol for the C-H cyanoalkylation of quinoxalin-2(1H)-ones (Scheme 1B).
Optimization studies began using 1-methylquinoxalin-2(1H)-one (1a) and cyclobutanoneO-(4-(trifluoromethyl)benzoyl) oxime(2a).In its final manifestation, an 80% isolated yield of 3-(3-cyanopropyl)−1-methyl-quinoxalin-2(1H)-one (3a) could be obtained under a constant-voltage of 3 V when graphite plates as the electrodes, DMA as the solvent, andnBu4NBF4(2.0 equiv.) as the electrolyte were applied (entry 1).At the outset, control experiments indicated that electricity (entry 2) and electrolyte (entry 3)were critical.BothnBu4NPF6andnBu4ClO4resulted in noticeable decreases in the overall reaction yield (entries 4 and 5).A solvent screen revealed that MeOH and DMF almost quenched the reaction (entries 6 and 7), while MeCN only marginally affected the yield (entry 8).Decreasing the amount ofnBu4NBF4or the amount of 2a lowered the yield (entries 9 and 10).Interestingly, constantvoltage of 3 V seemed to be significant for the reaction (entry 11).
Under the optimized conditions (Table 1, entry 1), we investigated the scope and generality of quinoxalin-2(1H)-ones (Scheme 2).Various substrates bearing electron-donating and electronwithdrawing groups (methyl, fluoro, chloro, bromo and trifluoromethyl) on the aromatic rings efficiently engaged in this reaction to afford the corresponding products 3b–3h in moderate to good yields.N-Unsubstituted quinoxalin-2(1H)-one was also successfully converted to the desired product 3i in 91% yield.Quinoxalin-2(1H)-ones with ethyl, butyl, allyl, propargyl, benzyl, ethoxycarbonylmethyl or benzoyl methyl groups on nitrogen were also suitable for this reaction (3j–3p).
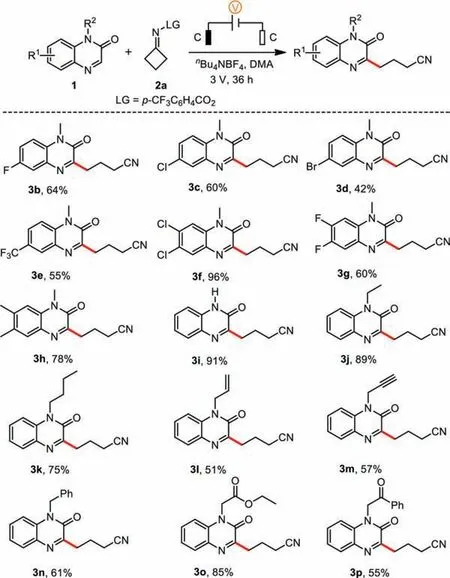
Scheme 2.Substrate scope of quinoxalin-2(1H)-ones. Reaction conditions: 1a(0.3 mmol), 2a (0.6 mmol), nBu4NBF4 (0.6 mmol), DMA (5 mL), undivided cell with two graphite electrodes (each 1.0×1.0 cm2), room temperature (r.t.), 3 V, 36 h.Isolated yields are reported.
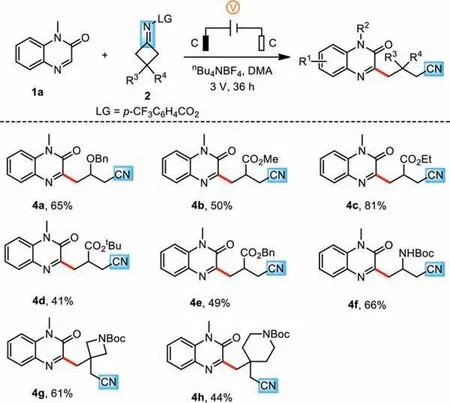
Scheme 3.Substrate scope of cyclobutanone oxime esters.Reaction conditions: 1a(0.3 mmol), 2a (0.6 mmol), nBu4NBF4 (0.6 mmol), DMA (5 mL), undivided cell with two graphite electrodes (each 1.0×1.0 cm2), room temperature (r.t.), 3 V, 36 h.Isolated yields are reported.
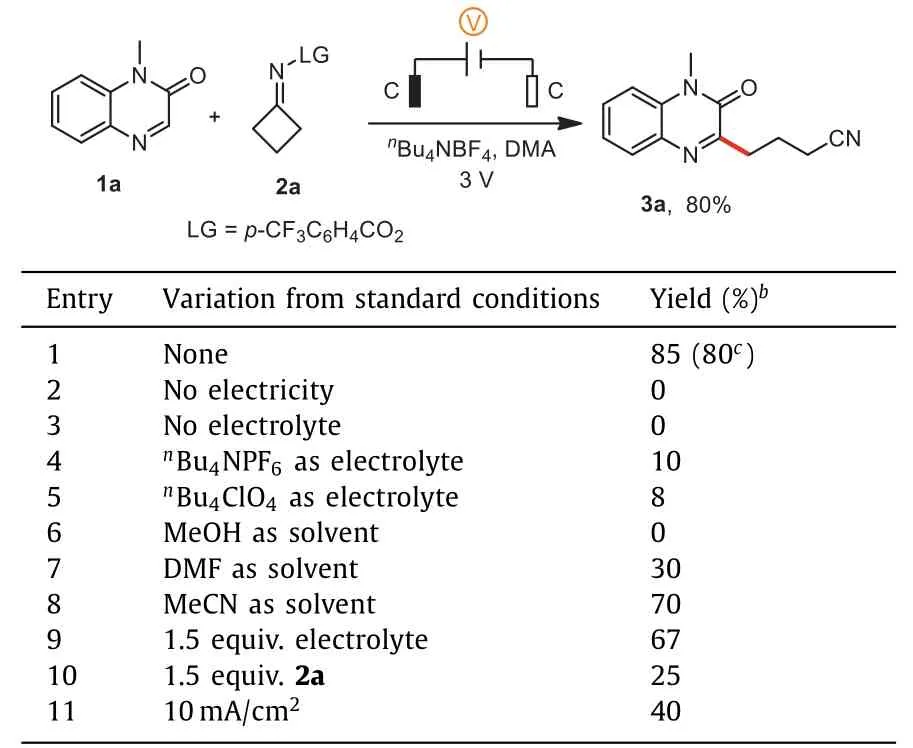
Table 1 Optimization of the reaction conditions.a
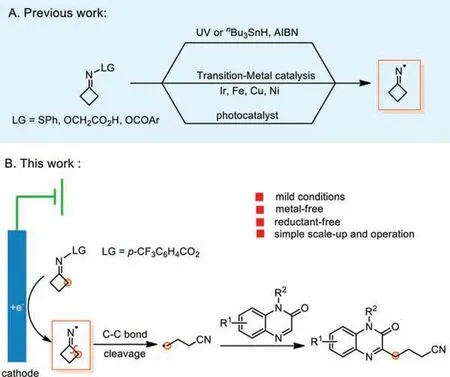
Scheme 1.Methods for redox of cyclobutanone oxime derivatives.
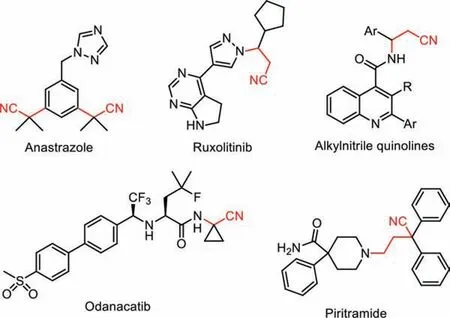
Fig.1.Representative pharmaceuticals containing alkylnitrile scaffold.
Next, we turned our attention to exploring the cyanoalkylation of quinoxalin-2(1H)-one 1a with various cyclobutanone oxime esters 2 (Scheme 3).A variety of cyclobutanone oxime esters containing benzyloxy and ester groups at the 3-position of cyclobutanone reacted well to give the target products 4a–4e in moderate yields.It is noteworthy that carbamate (NHBoc) also survived the reaction to yield the desired product 4f.Moreover, disubstituted substrates with large steric hindrance groups were capable of yielding 4g and 4h.
To demonstrate the utility and practicality of this electrochemical protocol, we used the device to perform a gram-scale reaction on a 5 mmol scale in 61% yield of 3a using the above-described conditions (Scheme 4).
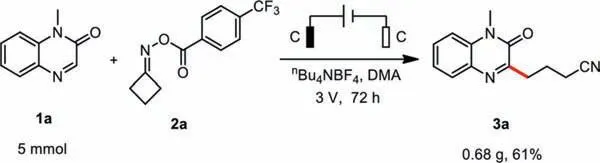
Scheme 4.Gram-scale of synthesis of 3a.
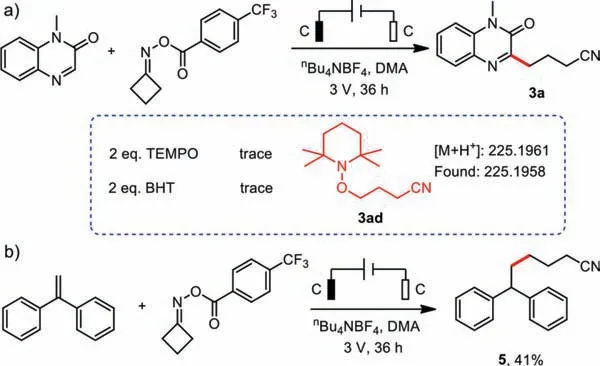
Scheme 5.Mechanistic studies.
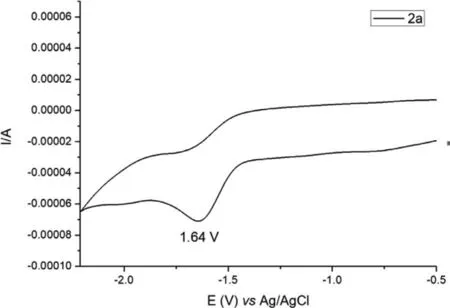
Fig.2.Cyclic voltammetry.
Several experiments were performed to gain insight into the reaction mechanism.Firstly, radical trapping experiments were conducted by the addition of (2,2,6,6-tetramethylpiperidin-1-yl)oxyl(TEMPO) and butylated hydroxytoluene (BHT) to the reactions of 1a and 2a, respectively (Scheme 5a).Both additives halted the reactions to some degree, and the radical trapping product 3ad was detected by high-resolution mass spectrometry.Secondly, when 1,1′-(1,2-ethenediyl) dibenzene was subjected to electrolysis in the presence of 2a under the standard conditions, a 41% isolated yield of the addition-compound 5 was obtained (Scheme 5b), which indicated that the cyanoalkyl radical was formed.Thirdly, the cyclic voltammetry showed that the reduction potential of substrate 2a was −1.64 V (vs.Ag/AgCl) (Fig.2), while reduction potential of the cathode was −1.75 V relatively, which means 2a can be reduced at the surface of the cathode under standard reaction condition.
On the basis of the above experimental observations, a plausible mechanism was proposed for the cyanoalkylation of quinoxalin-2(1H)-one (Scheme 6).The reaction begins with single-electron reduction of 2a at the cathode, affording iminyl radical I, which undergoes C−C bond cleavage to form cyanoalkyl radical II.Subsequently, the radical addition of II to 1a leads to radical intermediate III, which then undergoes single-electron oxidation at the anode followed by loss of H+to form product 3a.
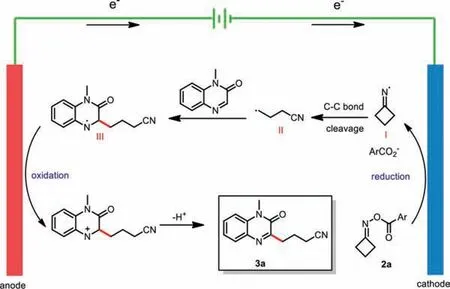
Scheme 6.Proposed mechanism.
In summary, a practical electro-reductive protocol for the direct C–H cyanoalkylation of quinoxalin-2(1H)-onesviaiminyl radical-mediated ring opening has been developed.A variety of quinoxalin-2(1H)-ones and cyclobutanone oxime esters can be applied in this environmentally benign reaction.Furthermore, it may provide a reasonable method for medicinal synthesis in the future.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We are grateful to the National Natural Science Foundation of China (Nos.21732002, 22077071) and Frontiers Science Center for New Organic Matter, Nankai University (No.63181206) for generous financial support for our programs.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.12.053.
 Chinese Chemical Letters2022年8期
Chinese Chemical Letters2022年8期
- Chinese Chemical Letters的其它文章
- Adsorptive removal of PPCPs from aqueous solution using carbon-based composites: A review
- A review on hollow fiber membrane module towards high separation efficiency: Process modeling in fouling perspective
- Recent advances in DNA glycosylase assays
- Chiral pillar[n]arenes: Conformation inversion, material preparation and applications
- Recent progress in carbon-based materials boosting electrochemical water splitting
- Working principle and application of photocatalytic optical fibers for the degradation and conversion of gaseous pollutants