
Two-dimensional Pt2P3 monolayer: A promising bifunctional electrocatalyst with different active sites for hydrogen evolution and CO2 reduction
2022-09-16 05:25YutingSunShungWngDongxuJioFengyuLiSiyoQiuZhongxuWngQinghiCiJingxingZhoChenghuSun
Chinese Chemical Letters 2022年8期
Yuting Sun, Shung Wng, Dongxu Jio, Fengyu Li, Siyo Qiu, Zhongxu Wng,Qinghi Ci, Jingxing Zho,∗, Chenghu Sun
a College of Chemistry and Chemical Engineering, Key Laboratory of Photonic and Electronic Bandgap Materials, Ministry of Education, Harbin Normal University, Harbin 150025, China
b School of Physical Science and Technology, Inner Mongolia University, Hohhot 010021, China
c School of Chemical Engineering and Energy Technology, Dongguan University of Technology, Dongguan 523808, China
d Department of Chemistry and Biotechnology, and Centre for Translational Atomaterials, Swinburne University of Technology, Hawthorn, VIC 3122, Australia
ABSTRACT Green hydrogen production and CO2 fixation have been identified as the fundamental techniques for sustainable economy.The open challenge is to develop high performance catalysts for hydrogen evolution reaction (HER) and CO2 electroreduction (CO2ER) to valuable chemicals.Under such context, this work reported computational efforts to design promising electrocatalyst for HER and CO2ER based on the swarm-intelligence algorithm.Among the family of transition-metal phosphides (TMPs), Pt2P3 monolayer has been identified as excellent bifunctional catalysts due to high stability, excellent conductivity and superior catalytic performance.Different from typical d-block catalysts, p-band center presented by P atoms within Pt2P3 monolayer plays the essential role for its reactivity towards HER and CO2ER, underlining the key value of p-electrons in advanced catalyst design and thus providing a promising strategy to further develop novel catalysts made of p-block elements for various energy applications.
Keywords:Bifunctional electrocatalysts HER/CO2ER Swarm-intelligence structure search Density functional theory Two-dimensional Monolayer
In recent decades, the over-consumption of conventional fossil fuels results in two severe problems: the energy crisis and global warming [1,2].Hydrogen (H2), which is regarded as the cleanest fuel, represents one of the most promising energy sources to alleviate the energy shortage by potentially reducing the fossil fuel dependence [3,4].H2production through electrochemical water splitting (also referred as hydrogen revolution reaction, HER) has been regarded as one of the most important technologies for the energy source of the next generation.On the other hand, CO2conversion has been widely considered as a promising strategy in alleviating the greenhouse effect and solving the environmental concern[5,6].More importantly, as an abundant C1feedstock, the reduction of CO2to high-value carbonaceous with improved energy density,such as methane (CH4) and formic acid (HCOOH), is extremely appealing for renewable energy storage, resulting in an eco-friendly carbon-neutral cycle [7,8].Among various methods for CO2reduction, the electroreduction of CO2(CO2ER) has attracted considerable attention owing to its ambient aqueous operation environment [9,10].In addition, it converts the intermittent electricity into stable chemical energy and generates valuable products, which can be readily controlled by changing the operating potential, the electrolyte, pH,etc.[11,12].
As well known, catalysts are the core components in catalysis.To date, Pt-based materials often serve as HER benchmark catalysts due to their high exchange current density and small Tafel slope [13], while the high cost and scarcity greatly hamper their widespread utilization.In recent years, although some non-Pt and metal-free HER catalysts have been reported, their catalytic activity and long-term durability are obscured by Pt-based catalysts [14].On the other hand, metal-based materials, such as (especially) Cu[15], Pd [16], Au [17], Ag [18] and Pt [19], were extensively utilized as the CO2ER catalysts, however, which were significantly hindered by their large overpotential, poor selectivity, high cost or low stability.Thus, developing highly efficient, low cost, and sustained catalysts for HER and CO2ER is paramount and apparently rewarding for the sustainable and large-scale implementation of clean energy devices.
Incorporating non-metal elements, such as phosphorus (P), sulfur (S) and boron (B) into metals to form multicomponent alloys is a promising strategy to eliminate the usage of these precious metal-based catalysts for cost-effective applications [20–24].In particular, the introduction of non-metal elements can effectively tailor the electronic structure and surface properties of metal catalysts, endowing them better catalytic performance.In this regard, as one of the most earth-abundant elements, P-based materials have been emerging as a kind of promising electrocatalysts due to their intrinsic electrochemical activity and widely tunable properties [25].
Recently, transition-metal phosphides (TMPs), with the formula of MxPy, have attracted immense attention as promising HER catalysts due to their hydrogenase-like catalytic mechanisms and good conductivity [26–33], in which the P atom plays a key role, since it not only performs as the active site for HER, but also tunes the electronic properties of metal sites with an appropriate ratio to boost the catalytic performance.Notably, P anions can also protect the TMPs from dissolving in the electrolyte, thus endowing TMPs a high durability during the electrocatalysis.In addition to HER, TMPs can also been widely applied in other energy-related electrocatalysis.For example, Li’s and coworkers reported that their synthesized rhodium phosphide (Rh2P) catalyst exhibits higher catalytic activity for water splitting [27].Kucernaket al.fabricated the PdP2and Pd5P2particles dispersed on carbon and found that the PdP2material showed good activity towards the oxygen reduction and formic acid oxidation [28].Sun’s group proposed that FeP nanoarray can be utilized as an efficient catalyst for CO2ER to methanol and alcohol [29].Although considerable efforts have been made to explore the potential of bulk TMPs as electrocatalysts, the precisely controllable design of specific function-oriented TMPs-based catalysts with tunable compositions, sizes, and morphology constraints is still challenging [34].Especially, the density of the exposed active sites is rather limited in bulk materials.Thus,the development of novel TMPs with uniform and specific exposed facets is essential to further boost their catalytic activity.Thus, designing novel TMPs-based catalysts with abundant exposed active sites, favorable structure and optimized chemical composition is highly desirable.
Since the discovery of grapheme in 2004 [35], two-dimensional(2D) materials have triggered increasing interest for developing efficient electrocatalysts [36–38].Compared with the traditional materials, 2D materials exhibit obvious structural advantages benefiting to their application in the field of catalysis, including high specific surface area and large surface atomic ratio.Inspired by the extensive studies of 2D materials, many 2D TMPs-based nanosheets have been reported experimentally and theoretically, which show interesting properties for various applications [39–48].For example, Yanget al.proposed a topochemical strategy to successfully synthesize 2D Co2P and Fe2P using phosphorene templates, which can perform as promising OER electrocatalysts due to the effective charge-transport modulation and improved surface exposure[39], while Sunet al.obtained high-quality 2D MnP single crystals on liquid metal Sn, which exhibit intrinsic ferromagnetism for the development of spintronic devices [40].Theoretically, Yoonet al.proposed that the layered Sr2P and Ba2P sheets are thermodynamically stable and could be applied as 2D electrides [41].In addition,Shaoet al.showed that 2D W2P and Fe2P monolayers exhibit high HER catalytic activity [42], whereas Chen’s group showed that the doped Mo2P monolayers can act as promising candidates for highperformance lithium-ion battery anode materials [43].
Having in mind the excellent properties and the great potential applications of 2D TMPs, in this work, we carried out a comprehensive swarm structural search to study if P alloying can alter the electronic structure of TMPs, in which the P-rich platinum phosphide was chosen as an example, because 1) Pt is the commonly-used element to form TMPs for energy conversion and storage, and 2) P-rich TMPs generally exhibit higher catalytic activity.Interestingly, we successfully identified a new Pt2P3monolayer as the electrocatalyst, which exhibits high stability and inherent metallic feature.More interestingly, differentpband-center reflected by the P atoms within Pt2P3monolayer endows this material excellent HER and CO2ER activity, which is different from the typical d-block catalyst.Our findings suggested that our proposed Pt2P3monolayer can serve as an ideal bifunctional catalyst for H2production and CO2conversion with superior catalytic performance, large surface area, good conductivity, and high stability.
The particle swarm optimization algorithm within the CALYPSO code [49], was employed to explore the atomic structure of the 2D Pt2P3monolayer, which can efficiently search for the ground or metastable structures just through the given chemical compositions.Especially, this method has successfully predicted many novel 2D materials with excellent properties and wide applications[50–55], among which some materials have been experimentally synthesized [56,57].Furthermore, the structural optimization and property computations were performed using the density functional theory (DFT) method, as implemented in the Viennaab initiosimulation package (VASP) [58,59], in which the ion–electron interaction was treated by the projector-augmented wave (PAW)method [60,61].We adopted the Perdew-Burke-Ernzerhof (PBE)functional within the generalized gradient approximation (GGA)for structural optimization [62], while the HSE06 functional was employed to compute the band structures and density of states[63].The cut-off energy for the plane-wave basis set was set to 550 eV with an energy precision of 10−5eV, and the atomic positions were fully relaxed until the force on each atom was less than 10−3eV/.
A 3 × 3 supercell was built to simulate the Pt2P3monolayer, in which a vacuum distance of 20in thez-direction was employed to avoid the interactions between adjacent layers.A 5 × 5 × 1Гcentered Monkhorst−Pack (MP) kpoint grid was adopted for the geometry relaxation and self-consistent calculations.The van der Waals (vdW) interactions were determined using the DFT + D3 correction [64].Dynamic stabilities and phonon dispersion curves were obtained according to the supercell approach within the Phonopy code [65].At the same time, first-principles molecular dynamics (MD) simulations were performed to evaluate the thermal stability of the predicted Pt2P3monolayer for 10 ps with a time step of 1.0 fs, in which the temperature was controlled by employing the Nosé-Hoover method [66].The free energy profiles, which are efficient in estimating the performance of electrocatalytic reactions, were determined by applying the computational electrode model (CHE, see Supporting information for the further computational details) [67,68].
After an extensive structure search (Figs.S1 and S2 in Supporting information), a hitherto unknown 2D Pt2P3monolayer with space groupCmwas achieved (Fig.1a, and its corresponding POSCAR file was presented in Supporting Information).The unit cell of Pt2P3monolayer with the lattice of 3.68, consists of two inequivalent Pt’s posistions (Pt1: 0.0609, 0.9759, 0.5838) and(Pt2: 0.6229, 0.6176, 0.4098) and three inequivalent P atoms occupying at the (P1: 0.4488, 0.2341, 0.6239), (P2: 0.6943, 0.5093,0.5235), and (P3: 0.9953, 0.8759, 0.3086).As a result, the coordination numbers of the two Pt atoms are different: the former is coordinated by six P atoms with the Pt−P bond length in the range of 2.36∼2.46, while the latter is tetracoordinated with four P atoms, leading to the formation of a tetra-sublayer structure with the thickness of ∼5.27(Fig.1b).On the other hand, the three P atoms are surrounded by three Pt atoms with Pt-P bond length ranging in 2.02∼2.61.Specially, P1and P2atoms are interconnected with each other, forming the quasi-square Pt2P2units and exhibiting covalent bonding character with the P1-P2bond length of 2.55.
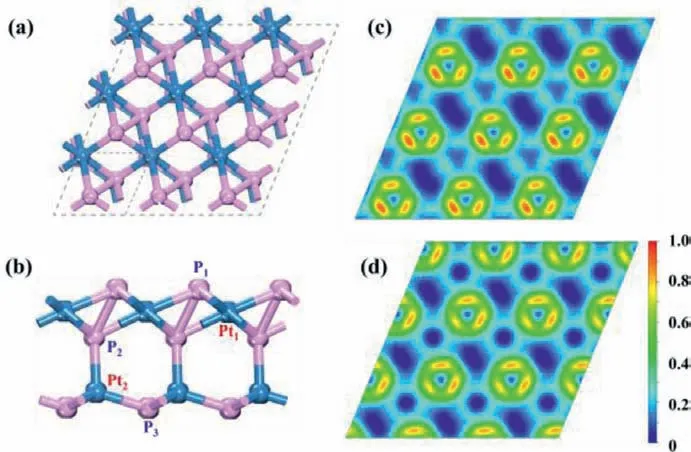
Fig.1.(a) Top and (b) side views of the predicted Pt2P3 monolayer.The unit cell is indicated by a gray dashed rhombus.Electron localization function (ELF) maps of a Pt2P3 monolayer along planes containing specified (c) Pt1−P1 and (d) Pt2−P3 bonds.
According to Hirshfeld charge analysis [69], P atoms can accept electrons from Pt, thus showing negative charge.Specifically, about 3.07 electrons are transferred from Pt to P atoms in the 3 × 3 supercell, and the Pt1, Pt2, P1, P2and P3carry 0.16, 0.18, −0.13,−0.12 and −0.09 electrons, respectively.Interestingly, the exposed P atoms with negative charges normally play an important role in the electrocatalytic reaction process.For example, P atoms in TMPs can perform as the active site to capture the positively charged H protons during electrochemical HER.Furthermore, we computed the electron localization function (ELF) to analyze the bond nature in Pt2P3monolayer, where a large ELF value (>0.5) represents the covalent bond or core electrons, while the ionic bond is obtained by a small ELF value (<0.5).As shown in Fig.1c and d, the Pt−P bond exhibits ionic bonding character, and the two nearest P atoms are obviously covalent bond.The coexistence of ionic and covalent bonds is beneficial to stabilize Pt2P3monolayer.
The material stability is the most fundamental and critical factor for its practical applications.To this end, we computed the cohesive energy of the Pt2P3monolayer (more computational details were presented in Supporting information), which is shown to be a well-accepted parameter to ascertain the feasibility of the experimental synthesis of the predicted 2D materials [58–64].Our results showed that the cohesive energy of the Pt2P3monolayer was computed to be 4.74 eV per atom, which is higher than those of the experimentally available phosphorene (3.30 eV per atom) [70] and silicene (3.98 eV per atom) [71], at the same level of theory.Thus,the predicted Pt2P3monolayer is quite likely to be synthesized under certain experimental conditions.
To examine the dynamical stability, phonon dispersion has been calculated with Phonopy code.No imaginary frequency was observed in the entire Brillouin zone (Fig.S1h), suggesting the dynamic stability of this 2D Pt2P3monolayer.Furthermore, AIMD simulations were performed at 500 K using a 3 × 3 supercell with a time step of 1 fs.As shown in Fig.S3 (Supporting information), the snapshots of the obtained structure indicated that Pt2P3monolayer can well maintain its structure, implying good thermal stability.Also, we assessed the mechanical property of Pt2P3monolayer by computing its elastic constants with the finite distortion method, with elastic constants C11= 88.21, C22= 94.05,C12= 49.79, and C44= 20.84 N/m, which satisfy the criteria of a mechanically stable 2D material (C11C22– C122>0 and C44>0)[72], indicating good mechanical stability.
Another more interesting fact is that the electrochemical stability of materials is also crucial for their practical applications in electrocatalysis, as the electrochemical reactions usually proceed under realistic electrochemical environments.We thus adopted the dissolution potential (Udis) to assess the electrochemical stability of the Pt2P3monolayer against dissolution under HER and CO2ER conditions (more computational details were summarized in Supporting information).Our results showed that the computed Udisvalue of Pt atom in Pt2P3monolayer is 2.38 V, which is much higher than the U0of 0.00 and 0.43 V for generating H2and HCOOH products (the main products for HER and CO2ER on Pt2P3monolayer, as discussed latter).Thus, we predicted that Pt2P3monolayer can survive under realistic experimental conditions, which could be ascribed to the strong binding strength between Pt and P atoms within 2D Pt2P3framework, thus suggesting its excellent electrochemical stability.
In general, the electronic property of catalyst strongly correlates with its activity in the electrochemical reaction, since good electrical conductivity can ensure the fast charge transfer.In this regard, we computed the electronic band structures and projected density of states (PDOS) of the Pt2P3monolayer using the HSE06 functional.Our results demonstrated that the Pt2P3monolayer exhibits intrinsic metallicity, as some band levels are crossing the Fermi level (Fig.2a).On the other hand, the PDOS peaks at Fermi levels are high, which are mainly composed of P-3p states, suggesting the available electrons that can participate the electronic transport and thus facilitating to its performance in electrocatalysis.In addition, there are obvious hybridizations between Pt-5d orbitals and P-3p, and P1–3p and P2–3p orbitals (Figs.2b-d), further suggested the strong chemical bonding between Pt atoms and P atoms, and two P atoms in the Pt2P3monolayer.Overall, these above results showed that the novel Pt2P3monolayer exhibits high stability and excellent electronic properties, thus holding great promise for its applications in electrocatalysis.
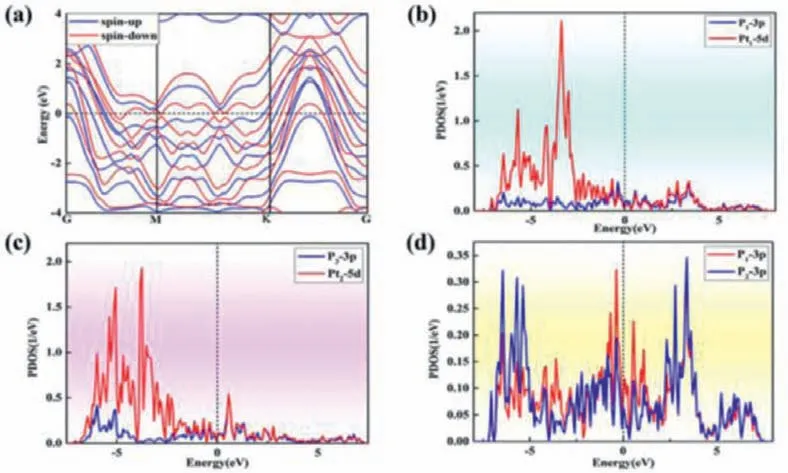
Fig.2.(a) The computed band structure and projected density of states (PDOSs) of (b) P1–3p and Pt1–5d, (c) P3–3p and Pt2–5d, and (d) P1–3p and P2–3p for Pt2P3 monolayer.The Fermi level was set to zero.
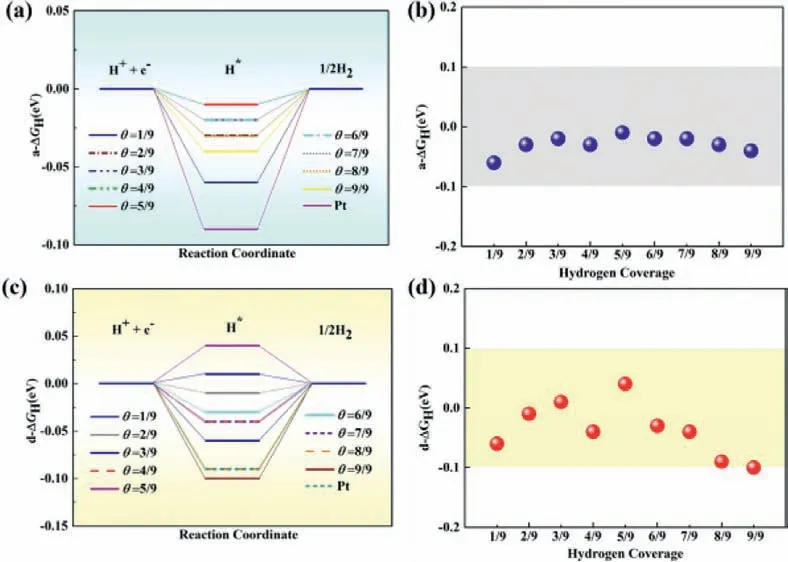
Fig.3.The computed average Gibbs free profiles and the corresponding free energy changes (a, b) a-ΔGH and (c, d) d-ΔGH in individual process as a function of H coverage(θ) on the Pt2P3 monolayer.The free energy window (± 0.1 eV) with catalytic activity comparable to that of Pt is highlighted in light area.
According to previous studies [73], TMPs-based materials have been widely reported as the promising catalysts for HER.Inspired by the unique structure, good stability, and outstanding electrical conductivity of the Pt2P3monolayer, we further explored its catalytic activity towards the HER.
In HER, a key descriptor to evaluate the activity of a catalyst is the Gibbs free energy of hydrogen adsorption (ΔGH), and the site withΔGHclose to zero is considered to be the active site for HER [74].To this end, we first examined the adsorption of the hydrogen atom on the Pt2P3monolayer by considering various adsorption sites.As expected, the negatively charged and exposed P atoms in the Pt2P3monolayer perform as the active sites to trap the positively charged H+protons due to the strong electrostatic interaction between each other, well consistent with the HER on other TMPs-based catalysts.Herein, both the average process (a-ΔGH) and individual process (d-ΔGH) were computed to assess the HER activity of the Pt2P3monolayer.In detailed, a-ΔGHrepresents the collective process, in which all H atoms on the surface are assumed to be simultaneously converted to molecules, whereas d-ΔGHwas employed to assess the individual process, in which H2molecule can be produced one by one.To explore the effect of hydrogen coverage on HER performance, a 3 × 3 × 1 supercell of the Pt2P3sheet was constructed for the adsorption ofnhydrogen atoms withnvarying from 1 to 9, corresponding to the hydrogen coverage (θ) ranges from 1/9 to 9/9.
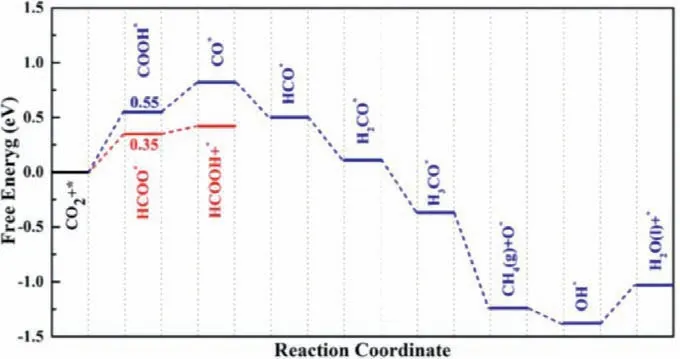
Fig.4.The free energy profile for CO2ER along various pathways on the Pt2P3 monolayer.
Due to the existence of two different exposed P atoms in the Pt2P3monolayer, namely, the P1and P3atoms (Fig.1b), we first examined the adsorption of one H atom (θ= 1/9) on the two P atoms.Interestingly, theΔGHvalues were computed to be −0.31 and −0.06 eV, respectively, on the P1and P3sites, suggesting that the latter will exhibit superior HER catalytic activity.Based on the above results, we studied the effect of hydrogen coverage on the HER activity of the Pt2P3monolayer, in which all H atoms were adsorbed on the side that the P3atoms are locating.Encouragingly, as shown in Figs.3a and b, the computed a-ΔGHvalues are −0.03, −0.02, −0.03, −0.01, −0.02, −0.02, −0.03 and−0.04 eV, respectively, atθ= 2/9, 3/9, 4/9, 5/9, 6/9, 7/9, 8/9 and 9/9, which are close to zero and comparable to that of Pt(−0.09 eV), further indicating the superior HER performance of the Pt2P3monolayer at whether low or high H coverage.In the individual process, however, d-ΔGHvalues are computed to be −0.01,0.01, −0.04, 0.04, −0.03, −0.04, −0.09 and −0.10 eV, respectively,atθfrom 2/9 to 9/9 (Figs.3c and d).Thus, at lower H coverages (θfrom 1/9 to 7/9), the collective and individual processes can simultaneously take place, and at higher H coverages (θ= 8/9 and 9/9),the collective process exhibits higher catalytic activity, because itsΔGHvalues is closer to 0.
In the CO2ER, the first step is the hydrogenation of CO2molecule to HCOO∗or COOH∗intermediates.To this end, we examined the adsorption of the two species at various sites, including the exposed P1, P3, and their adjacent Pt atoms.Our results showed that HCOO∗and COOH∗intermediates are energetically favorable to adsorb on the P1site with the adsorption energies of−2.95 and −2.44 eV (Fig.S4 in Supporting information), respectively, which are slightly larger than those of on the P3site (−2.77 and −2.24 eV), indicating that P1atom will perform as the active site for CO2ER.
Next, we studied the CO2ER mechanism on the P1site of the Pt2P3monolayer (Fig.4), and the corresponding intermediates were presented in Fig.S5 (Supporting information).According to the computed free energy profiles (Fig.4), we found that the formation of HCOO∗species is uphill by 0.35 eV, which is smaller than that of COOH∗(0.55 eV), implying that CO2molecule is energetically favorable to be hydrogenated to the HCOO∗species, as compared with COOH∗one.Subsequently, the approaching of the second (H++ e−) to the HCOO∗yields the HCOOH product with theΔGof 0.08 eV.During the CO2ER to HCOOH product, the potential-determining step (PDS) locates at the first hydrogenation step (CO2→HCOO∗) due to itsΔGvalue (0.35 eV).Thus,the computed limiting potential (UL) for CO2ER on the designed Pt2P3monolayer is −0.35 V, which is comparable (even less negative) to those of some traditional metal-based benchmark (−0.19∼−0.60 V) [75–78], implying its superior catalytic performance for CO2reduction to HCOOH product.
In addition, we also explored the product distribution for CO2ER on the Pt2P3monolayer by examining the reaction pathway along the COOH∗hydrogenation.As shown in Fig.4, we found that the approach of a second hydrogen induces the dissociation of COOH∗into CO∗and H2O, which is mildly endothermic by 0.27 eV.The CO∗is further hydrogenated to HCO∗, H2CO∗, H3CO∗, and the final product CH4, with the computedΔGvalues of −0.32, −0.39,−0.48 and −0.87 eV, respectively.Notably, the remaining O atom on the P1active site can be further reduced to OH∗and H2O(ΔG= −0.14 and 0.36 eV).Specially, in this process for CO2ER to CH4production, the COOH∗formation is identified as the PDS owing to its maximumΔGvalue of 0.55 eV, corresponding to the ULof −0.55 V.Clearly, HCOOH production is thermodynamically favorable by 0.20 eV relative to CH4production.To estimate the distribution of different products (HCOOHvs.CH4), the thermodynamic formula exp[−(ΔG)/(RT)] can be employed according to their free energy difference, in which the values ofΔGandTare 0.20 eV and 291.65 K.Thus, the HCOOH:CH4molar ratio is about 2000:1 at ambient temperature, suggesting a strong selectivity toward the HCOOH product.
Finally, it is important to consider the completion between CO2ER and HER on the P1active site, since the HER might affect the Faradaic efficiency significantly.To this end, we computed the difference between the limiting potentials for CO2ER and HER(ULCO2ER– ULHER) to assess the catalytic selectivity of P1site towards the two reactions.According to previous reports [79], a less negative value of (ULCO2ER– ULHER) represents a higher selectivity towards the CO2ER.Interestingly, the (ULCO2ER– ULHER) value on P1site is computed to be only −0.04 V, which is much less negative than that of metal-based benchmark (−0.50 V), suggesting the excellent selectivity of the P1active site towards the CO2ER.
Inspired by the superior bifunctional activity of the Pt2P3monolayer, we further accessed its viability in other TMPs with stoichiometry of TM2P3, in which Fe, Co, Ni, Mo, Ru, Rh and Pd were chosen to substitute the Pt atom in Pt2P3monolayer, as they are the most frequently employed elements among TMPs.After fully geometrical optimizations, we found that similar twodimensional structures can be obtained in TM2P3monolayers for TM = Fe, Co, Ni, Mo, Ru, Rh and Pd.Unfortunately, these systems are dynamically unstable due to imaginary frequencies in phonon dispersions (Fig.S1), thus highlighting the special role of Pt in stabilization of the novel 2D structure.
Similarly, we also tried to search for other Pt-based TMPs, including PtP, PtP2and PtP3monolayers.Our results showed that significant imaginary vibrational frequencies can be observed in their computed phonon dispersion curves, thus indicating their dynamical instability, whereas the PtP3monolayer is dynamically stable due to the absence of imaginary frequencies.Yet, the structure of the PtP3monolayer will be greatly distorted and reconstructed after performing AIMD computations by annealing at 300 K for 1 ps with a time step of 1 fs, suggesting that the predicted PtP3monolayer is thermally unstable at ambient conditions.In addition,the morphology of a given catalyst may affect its catalytic activity.Thus, we took the Pt2P3bilayer (Fig.S6 in Supporting information)as an example to examine the effect of the morphology on the catalytic activity of Pt2P3material toward HER and CO2ER.Our results showed that the limiting potentials for the two reactions on P1and P3sites of Pt2P3bilayer are computed to be −0.64 and −0.36 eV,which are more negative than those of on Pt2P3monolayer, indicating that the stacking of two Pt2P3monolayers will hamper its catalytic performance.Also, we noted that no prior study was reported on the experimental synthesis of our proposed Pt2P3monolayer to date.However, recently, Yanget al.proposed a general bottom-up topochemical method to synthesize a series of 2D metal phosphides, such as Co2P, Ni12P5and CoxFe2-xP by using phosphorene sheets as the phosphorus precursors and 2D templates [39],which exhibited efficient electrocatalytic ability for OER.This pioneering study could open a promising strategy to synthesize Pt2P3monolayer by adopting similar experimental procedure in the near future, in which 2D phosphorene and soluble PtCl4coordination compounds may be employed phosphorus and Pt precursors, respectively.
As demonstrated above, Pt2P3monolayer exhibits excellent catalytic performance for HER and CO2ER, which can proceed on two different sides.Then, an interesting question naturally arises: why do different sides of the Pt2P3monolayer exhibit different catalytic activity, especially for HER? To gain a profound understanding on this question, the projected density of states (PDOSs) of different active sites were analyzed, since the chemical activity of a given catalyst is generally dependent on its underlying electronic properties.The PDOSs of various active sites, including P1and P3within the Pt2P3monolayer, were presented in Fig.S7 (Supporting information), where the 3p-PDOSs of P sites were mainly examined.To elucidate the remarkable difference between the adsorption strength of H∗species on the P1and P3sites, we evaluated their energy levels of the p-band center of (ɛp) model as [80–86]:ED(E)dE/D(E)dE, whereD(E) is the density of states of p band of the P site at a given energyE, and the Fermi level is set to zero.Our results demonstrated that theɛpvalues for P1and P3sites are computed to be −0.86 and −1.58 eV, respectively.Obviously, the position of theɛpfor P1site closer to the Fermi level can induce antibonding states to a higher energy, normally making them more difficult to be filled and thus resulting in a stronger H∗adsorption on the P1active site (ΔG= −0.31 eV).For P3site, however, its smallerɛpvalue indicates a relatively weak interaction of H∗species, which is responsible for the good catalytic performance for the HER on P3site.Similarly, the formed HCOO∗species during CO2ER prefers to adsorb the P1active site due to its less negativeɛpvalue (−0.86 eV), leading to its high catalytic performance for CO2ER.
In summary, by performing first-principles swarm-intelligence structural search computations, we predicted a hitherto unknown Pt2P3monolayer with metallic nature.Remarkably, the high cohesive energy, excellent thermal and dynamic stability, and outstanding mechanical properties of the Pt2P3monolayer, originating from its unique bonding arrangement,i.e., the coexistence of Pt−P ionic bond and p−p covalent bond, render it great promise for experimental synthesis.Interestingly, the different P sites on the two sides of the Pt2P3monolayer endow its ultra-high catalytic activity towards HER and CO2ER to HCOOH product, which can be mainly ascribed to their differences in p-band center, thus suggesting that the as-designed Pt2P3monolayer can perform as a promising bifunctional catalyst for H2production and CO2conversion.Our results are encouraging further experimental and theoretical research to explore novel 2D materials with appealing properties and potential applications in catalysis.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was financially supported by the Natural Science Funds for Distinguished Young Scholar of Heilongjiang Province(No.JC2018004), the National Natural Science Foundation of China(No.11964024), the “Grassland Talents” project of Inner Mongolia autonomous region (No.12000-12102613), and the Young science and technology talents cultivation project of Inner Mongolia University (No.21221505).We also thank the computational resource supported by Harbin Normal University and Beijing Paratera Technology Co., Ltd.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.11.034.
 Chinese Chemical Letters2022年8期
Chinese Chemical Letters2022年8期
- Chinese Chemical Letters的其它文章
- Adsorptive removal of PPCPs from aqueous solution using carbon-based composites: A review
- A review on hollow fiber membrane module towards high separation efficiency: Process modeling in fouling perspective
- Recent advances in DNA glycosylase assays
- Chiral pillar[n]arenes: Conformation inversion, material preparation and applications
- Recent progress in carbon-based materials boosting electrochemical water splitting
- Working principle and application of photocatalytic optical fibers for the degradation and conversion of gaseous pollutants