
具有制氢性能的镍基分子催化剂的密度泛函研究
2022-09-16 08:52武海素石海霞苗体方
淮北师范大学学报(自然科学版) 2022年3期
黄 颍,武海素,石海霞,李 双,苗体方
(淮北师范大学 化学与材料科学学院,安徽 淮北 235000)
0 引言
工业的快速发展和化石能源的大量消耗导致一系列的环境污染问题,同时,人口的迅猛增加和与之不相匹配的能源短缺矛盾变得日益尖锐,并深深制约着社会的发展[1].H2凭借高热值、无污染以及能储存的优良性能受到一致的好评并得到广泛的研究.遗憾的是,在全球氢气供应中96%的氢气来自于石油和天然气的蒸汽重整,这严重违背不依赖化石燃料储备的初衷.而其余4%的氢气则是通过电解水制得的,这种方法在获得氢能的同时损耗一定的电能,也难以彻底摆脱化石燃料对其的束缚.因此,光催化分解水制氢成为最理想和有效的方式将取之不尽的太阳能转换为氢能.
当前阶段,已经有多种分子催化剂被报道用于在水溶液中催化制氢,例如:钌催化剂[2]、铂催化剂[3]等,尽管它们具有较好的催化活性和较长的使用寿命,但高昂的成本迫使人们不得不寻找地球上廉价的金属去替代贵金属完成催化体系的构建.基于此,一系列廉价的金属催化剂被设计开发出来,例如:铁配合物[4]、钴配合物[5]、镍配合物[6]等.值得注意的是,金属镍凭借超低的成本和优良的催化性能受到研究者的极大青睐.廉价的分子催化剂搭配光敏剂和电子牺牲剂构成一个标准的三组分反应体系,遗憾的是,扮演重要角色的光敏剂往往是由一些昂贵的金属(铂、金、钯等)组成,且需要额外添加有机试剂作为反应中的H+来源.贵金属的引入和有机试剂的添加不仅造成环境的二次污染还加大成本的投入,达不到廉价高效的催化制氢目的.基于此,开发出在水溶液中具有催化制氢性能的廉价分子催化剂就变的至关重要.
除此之外,实验法通常是开发高效催化剂的常用方法,但是它耗时长、并且需要耗费巨大的人力和物力[7].为提高催化剂开发的效率,密度泛函理论(DFT)的引入对于研究催化剂的结构和制氢活性起着重要作用[8],其优势在于可以在催化剂合成之前对催化剂的制氢性能进行预测,根据预测的结果进行选择性合成,这样能避免盲目合成,有效提高开发催化剂的效率.在本文中,通过DFT深入研究2种具有不同空间结构的镍配合物,其结构示意图及一些原子编号,见图1,分析不同的取代基对其结构、吸收光能力、前线分子轨道、静电势及平均离子局部化能的影响,探究催化剂的制氢活性及结构的关联性,为后续催化剂的开发和设计提供理论依据.
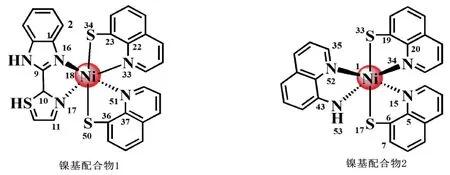
图1 镍基配合物1和2的结构示意图和原子编号(不显示氢原子)
1 计算方法
本文是在UB3LYP/6-31G(d)水平上,采用密度泛函理论对化合物进行优化[9].以优化的几何结构为基础,并用频率计算,计算结果表明,所有频率都是正值,表明经过优化而得到的2个化合物的几何结构都是稳定结构.在获得的结构基础上,采用含时密度泛函理论(TDDFT)方法,计算100个单重激发态,模拟出电子吸收光谱图.此外,在此结构基础上,计算出这2个化合物的静电势、平均离子局部化能及前线分子轨道,并模拟出化合物的最高占据轨道(HOMO)和最低未占据轨道(LUMO)的分子轨道图.本文中的轨道图、静电势和平均离子局部化能均通过Multiwfn程序绘制[10],所有的计算由Gaussian 16完成[11].
2 结果与讨论
2.1 结构分析
配合物1和配合物2都是以镍为中心原子的配合物,它们都可以作为光催化制氢的催化剂,由于取代基的不同使得这2个分子结构发生变化,进而影响催化剂的催化效果.本文通过密度泛函理论对2个化合物进行优化,得到2个化合物的稳定结构.计算得到的几何参数见表1.
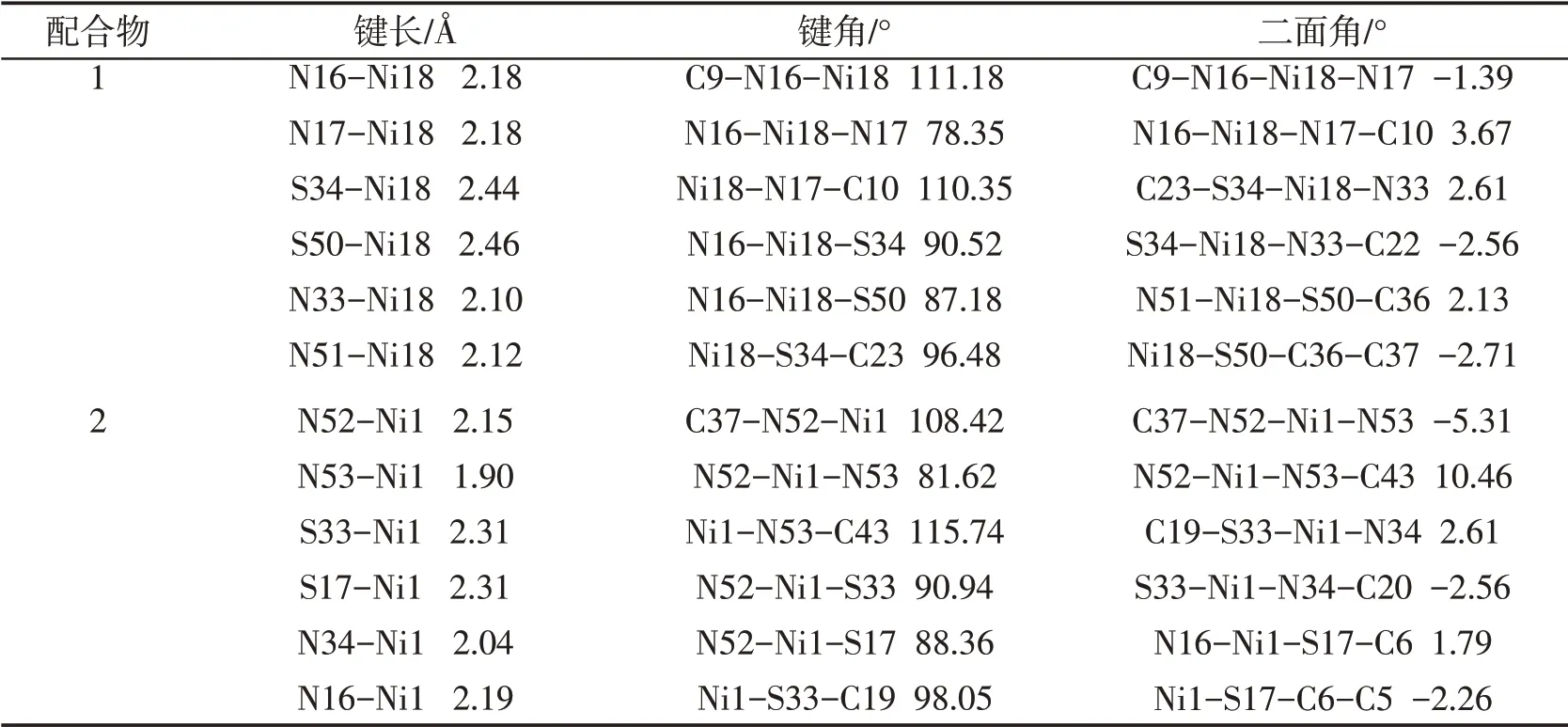
表1 配合物1和2结构的主要参数
从表1所得的数据可以看出,键长总体上变化不大,而键角、二面角之间差别较为明显,且二面角大小有正有负.例如配合物1中的S34-Ni18的键长2.44Å,对应的配合物2处S33-Ni1键长仅有2.31Å.键长越长,越容易断裂并与水溶液中的H+发生质子化反应.除此以外,二者的键角也有着较大的差别,化合物1的键角C9-N16-Ni18为111.18°,而配合物2的键角C37-N52-Ni1为108.42°.值得注意的是,二面角也有较大的变化.总而言之,配合物1相对于配合物2,有较光滑的平面可能会提高其在水溶液中溶解度,并暴露更多的活性位点,从而展现出更佳的催化制氢活性.
2.2 前线轨道分析
在优化的几何结构基础上,对分子轨道进行计算,得到化合物的最高占据轨道(HOMO)和最低占据轨道(LUMO),并模拟出配合物的HOMO和LUMO图,见图2.配合物1的151α-LUMO表明所有的电子云主要集中在左边的配体上,而150α-HOMO显示大多数电子云分布在上面的8-喹啉硫醇上,少量的电子分布在中心金属镍上.而对应的150β-LUMO,电子云分布在下面的8-喹啉硫醇上,同时有少量的电子云在左边的配体上,对于HOMO轨道,电子云主要分布在上面的8-喹啉硫醇上.对于配合物2来说,α自旋的LUMO轨道电子云主要分布在中心金属和连接镍的硫原子上.β自旋的轨道和配合物1基本上没有太大的差别.综上所述,尽管2种配合物的中心金属相同,由于配体的给电子差异,导致电子云的分布有较大的差异,无论配合物1还是2,当电子从HOMO轨道跃迁到LUMO轨道时,硫原子都能分布较多的电子云,这样有利于硫原子和周围的氢离子结合而释放氢气[12],说明这2个配合物都有一定的制氢活性.这些研究表明配体和官能团的改变对制氢活性有一定的影响,对于设计高性能催化剂具有一定的理论参考价值.
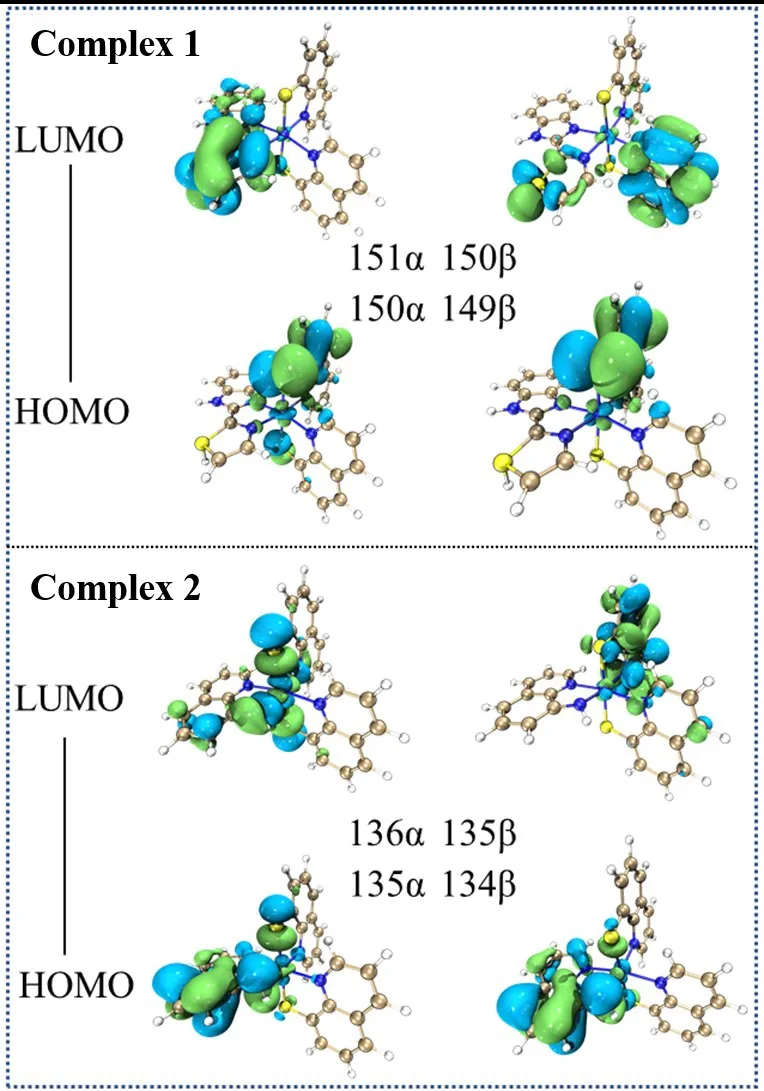
图2 配合物1和2的HOMO和LUMO轨道
静电势对于考察分子间静电相互作用、预测反应位点和预测分子性质等方面有重要意义.图3绘制了配合物1和2的静电势及静电势的极值点.其中,配合物1和2的静电势极小点分别为-41.76和-12.96 a.u..图4中配合物1和2的平均离子局部化能表明静电势的最小位置均出现在硫原子和中心金属镍原子附近.一般来说,中心区域部分表明电势低的位置,很容易发生亲核反应[12].上述结果表明2种配合物的活性位点均位于S和Ni原子处,与报道的类似配合物上的活性点一致[13].由于配合物1有较低的静电势,并且在LUMO上S和Ni原子有分布较多的电子云,配合物1应该具有较强的吸收氢质子的能力,理论上预测配合物1具有较好的制氢活性,目前配合物1和2正在合成中,其制氢活性很快可以通过实验测试得到验证.
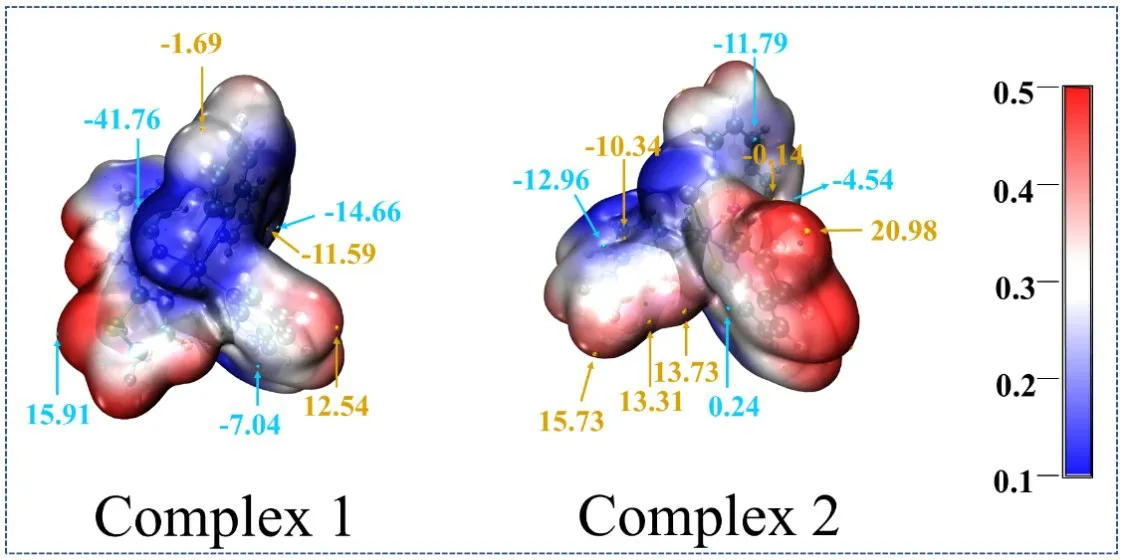
图3 配合物1和2的静电势(所有的单位均为a.u.)
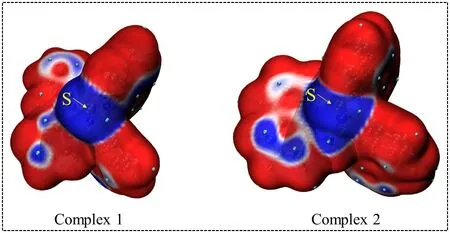
图4 配合物1和2的平均离子局部化能
2.3 紫外可见吸收光谱分析
从图5可知,配合物1和2分别在350 nm和290 nm处都有较强的吸收,同时在450 nm和495 nm左右也表现出光吸收能力,2个物质在可见光区域内展现出不同的光吸收能力,可能是由于配体的差异导致不同位置电子密度有所偏差,进而影响着吸收峰的位置[14].值得注意的是,尽管2个配合物在可见光区域内均展现出一定的光响应能力,但在实验方法制氢过程中,需要考虑光催化剂、光敏剂及电子牺牲剂的作用,尤其是光敏剂作为体系中吸收可见光并产生光生电子的重要物质,光敏剂和光催化剂镍配合物的相互作用也是要考虑的重要因素[15-17],有关光敏剂和光催化剂的相互作用研究,将在下一个工作中进行.
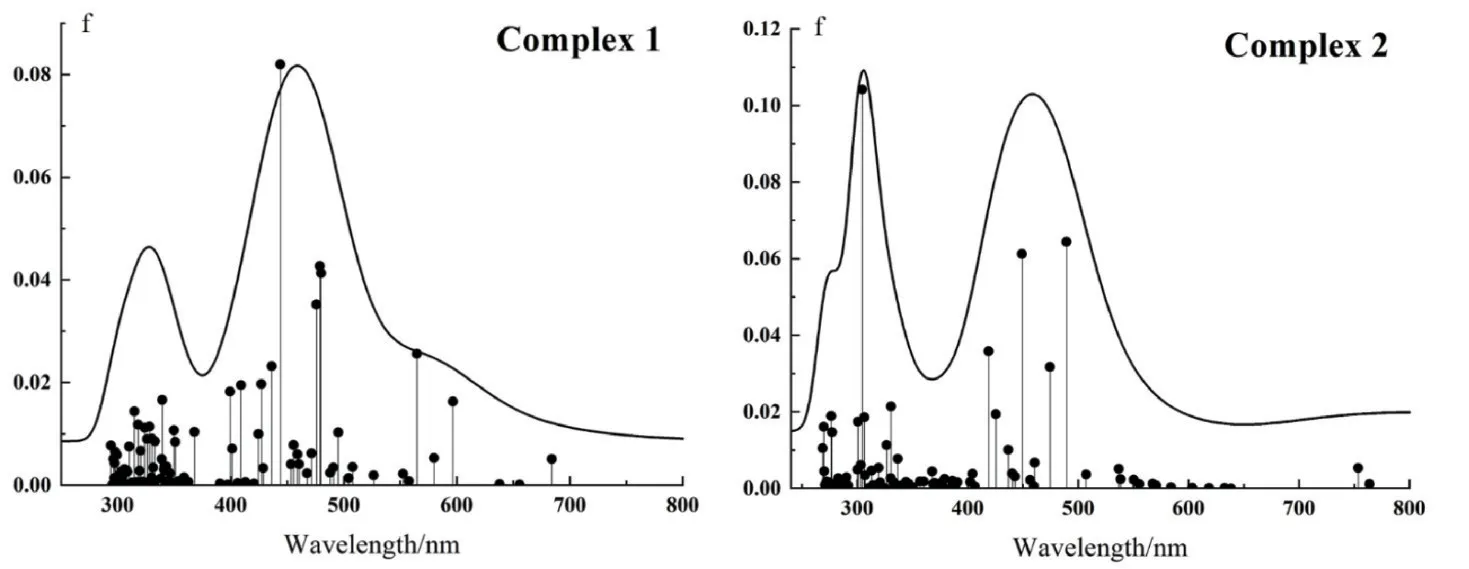
图5 配合物1和2的UV-Vis
3 结论
本文采用DFT方法,对2个镍基配合物进行理论计算,获得2个配合物的稳定结构,计算这2个配合物的前线分子轨道、静电势、平均离子局部化能及电子吸收光谱,探索配合物结构和制氢活性的关系,从理论上预测配合物1的光催化释氢性能要比配合物2更优秀,这些研究结果为设计、合成高性能光催化剂及探索光催化反应提供理论依据.
猜你喜欢
化工管理(2022年14期)2022-12-02
分子催化(2022年1期)2022-11-02
高等学校化学学报(2022年10期)2022-10-14
医学综述(2022年13期)2022-08-10
建材发展导向(2021年16期)2021-10-12
煤气与热力(2021年6期)2021-07-28
中国食物与营养(2020年12期)2020-09-10
上海建材(2020年12期)2020-04-13
表面工程与再制造(2019年6期)2019-08-24
智富时代(2018年3期)2018-06-11
