
氮磷掺杂方式对氧化石墨烯催化氧还原制H2O2性能的影响
2022-09-15 12:26:16孙泽涵刘祥瑞王昭晖陈咏梅
化学研究 2022年5期
孙泽涵,刘祥瑞,王昭晖,陈咏梅
(北京化工大学 化学学院,北京100029)
过氧化氢(H2O2)作为氧化剂使用时产物是水,无二次污染问题,因此广泛应用于化工、医药、环保、造纸、纺织、环保、航天及电子等领域[1]。现行的规模化生产工艺是蒽醌法,该工艺将烷基蒽醌与有机溶剂配制成工作溶液,在压力为0.30 MPa、温度55~65 ℃、有催化剂存在的条件下通入氢气进行氢化,再在40~44 ℃下与空气(或氧气)进行逆流氧化,经萃取、再生、精制与浓缩制得质量分数20%~30%的过氧化氢水溶液产品,目前市场上95%以上过氧化氢产品都是通过蒽醌法获得的[2]。然而,由于过氧化氢属于危化品,在运输、储存等方面存在着安全性风险[3-4];而另一方面,低浓度(质量分数2%~3%)过氧化氢水溶液即可用于日常生活和临床医用消毒。为此,基于氧气在阴极发生2电子还原过程(2e-ORR)设计而成的小型电化学装置可以用于现场制备H2O2水溶液[5-9],从而满足临床医学消毒等场合需求。
许多研究结果都证实O2分子在阴极表面发生2e-ORR过程可以生成H2O2,而O2分子在阴极表面发生4e-ORR过程生成OH-/H2O实际上是热力学更有利的过程,是制备H2O2电流效率的主要影响因素。研究表明,O2分子在阴极表面的还原过程受其在电极表面的吸附模式及结合能大小、导电性、多相催化等因素有关[10-11]。Norskov等通过理论计算认为,碳基材料表面OOH*吸附态较好的脱附能力是保证较高的2e-ORR选择性的关键[12-13]。众多研究者尝试对碳材料进行改性,通过在碳骨架中掺入杂原子而改变材料的电子结构,从而改变含氧中间体在材料表面的吸附状态,最终提高电催化制备过氧化氢的选择性[14-15]。
综合已有研究报道可知,掺杂在碳材料中的N原子主要有吡咯N、吡啶N和石墨N等几种形式[16-19],不同掺杂形式的N元素对碳材料2e-ORR的催化性能不同。理论计算和实验结果证实:吡啶N具有较好的O2吸附活性,但对OOH*中间态的吸附能力过强,因而更有利于发生4e-ORR过程;吡咯N的带隙较小,对OOH*的吸附能力适中,因而表现很高的2e-ORR反应活性;而石墨N附近的C原子对OOH*中间体的吸附能增强,因而也可以提高电化学制过氧化氢的性能。氮磷共掺杂碳材料的研究结果表明过多引入P原子会降低材料2e-ORR的性能,但是少量P原子的存在会促进吡咯N的形成,从而提高了2e-ORR的选择性[20-23]。
综上所述,N、P元素的存在形式对于氧气分子在碳材料表面的电化学行为有密切关联,然而其中的作用机理尚不明确。本研究基于水热法以尿素作为氮源、磷酸作为磷源对氧化石墨烯(GO)进行改性,考察了不同掺杂方式(共掺杂/顺序掺杂、掺杂比例等)所得到的改性GO的结构、电化学性能和电解制备过氧化氢的效果,藉此系统讨论了碳材料中N、P元素的存在形式对其2e-ORR性能以及生成H2O2电流效率之间的联系。
1 实验部分
1.1 仪器与试剂
氧化石墨烯,AR,深圳穗衡石墨烯科技;磷酸,AR,北京化工厂;尿素,AR,北京化工厂;5% Nafion溶液,杜邦DuPont;N,N二甲基甲酰胺(DMF),AR,天津市大茂化学试剂厂;碳纸,日本东丽公司;氢氧化钠,AR,上海阿拉丁生化科技股份有限公司;高锰酸钾,AR,北京化工厂。
电化学工作站,CHI760E,上海辰华仪器有限公司;数控超声波清洗器,KQ3200DE,昆山市超声仪器有限公司;电热恒温干燥箱,DHG-9053A,上海浦东荣丰科学仪器有限公司;K-Alpha 光电子能谱仪,美国Thermo Scientific公司;MSR电极旋转装置,AFMSRCE,美国PINE公司;RRDE电极,RRDEGC1156,天津艾达恒晟科技发展有限公司。
1.2 催化剂的制备
1.2.1 氮磷共掺杂氧化石墨烯的制备
100 mg GO、4.225 g H3PO4(0.043 mol)和3.480 g CO(NH2)2(0.058 mol)共混于50.00 mL去离子水中充分搅拌后,在室温下静置4 d。将浸渍完成后的悬浊液经超声分散后转移至水热釜中,放入烘箱150 ℃维持7.5 h,反应结束后自然冷却至室温。砂芯漏斗减压过滤后,用20 mL 0.2 mol/L NaOH和去离子水各洗涤3次。其后在60 ℃恒温干燥,得到产物,记作N1P0.37-GO。
采用相同的操作步骤,其中H3PO4用量为6.762 g(0.069 mol)、CO(NH2)2用量为1.200 g(0.020 mol),所得产物,记作N1P1.7-GO。
1.2.2 氮磷顺序掺杂氧化石墨烯的制备
100 mg GO和1.200 g CO(NH2)2(0.020 mol)混合于50.00 mL去离子水中,超声分散后转移至水热釜中,150 ℃下恒温7.5 h,反应结束后自然冷却至室温。砂芯漏斗减压过滤后,用20.00 mL去离子水洗涤3次, 60 ℃下干燥,得到的固体与6.762 g H3PO4(0.069 mol)混合如上进行水热反应,过滤后用20.00 mL 0.2 mol/L NaOH和去离子水各洗涤3次。60 ℃干燥后得到产物,记作N1-P1.7-GO。
1.3 结构表征
X射线光电子能谱(XPS)测量使用Thermo Scientific K-Alpha 光电子能谱仪,Al Kα射线(1 486.6 eV),本底真空度~3×10-7mbar,峰面积通过Lorentz(30%)和Gaussian(70%)函数拟合。数据处理采用XPSPEAK41软件进行曲线拟合。
1.4 电化学性能研究
使用MSR电极旋转装置和CHI760E双恒电位电化学工作站进行RRDE测试。所采用的旋转环盘电极:玻碳盘电极(直径为5.61 mm),铂环电极(外径7.92 mm、内径6.25 mm),理论电流收集率为37%;参比电极为Ag/AgCl(饱和KCl)电极,对电极铂片(1.0 cm×1.0 cm)电极。测试前依次用粒径为1 μm、0.3 μm、50 nm的Al2O3粉末打磨环盘电极,随后在含有10 mmol/L K3[Fe(CN)6]的1 mol/L KCl溶液中进行电流收集率的测试,测试时电极转速为1 600 r/min,电位扫描速率为50 mV/s。
催化剂负载:称取5 mg催化剂,加入950 μL DMF和50 μL Nafion溶液(质量分数5%),超声分散1 h得到催化剂分散液;在500 r/min的电极转速下,将50 μL催化剂分散液滴到玻碳盘上,然后在红外灯下旋干。
极化曲线的测定:0.1 mol/L KOH溶液中预先通30 min氧气,盘电位从1.0 V扫描到0.2 V(vs RHE,下同),扫速为5 mV/s;环电位固定为1.3 V。首先在1 600 r/min的电极转速下扫描两次,以活化电极并稳定背景电流;然后分别在400、625、900、1 225、1 600、2 025、2 500 r/min的电极转速下测试极化曲线,并使用Koutechy-Levich方程计算转移电子数。
1.5 电解实验
将0.2 mL质量分数5% 的Nafion溶液、3 mL N,N-二甲基甲酰胺(DMF)和1.8 mL去离子水混合制成分散液。取5 mg上述改性GO与0.5 mL分散液混合,超声分散均匀后喷涂于2.5 cm×2.5 cm的碳纸表面,在空气中用红外灯烘干电极上的分散液,制得阴极气体扩散电极。
电解实验采取三电极体系:所制得的气体扩散电极为工作电极,铂碳(Pt/C)电极作对电极,以饱和甘汞电极作参比电极,在通入恒流氧气的条件下,在2 mol/L NaOH溶液中进行-1.80 V恒电位电解1 200 s。
由测得的密度求得电解后阴极电解液的体积VH2O2。采用KMnO4滴定法测定电解液中的H2O2浓度,如下式计算生成过氧化氢的电流效率η:
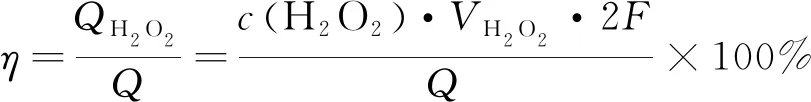
(1)
其中Q为电化学工作站记录的电解过程实际消耗电量。
2 结果与讨论
2.1 改性GO的结构表征结果
采用氮磷共掺杂方法所获得N1P0.37-GO样品的XPS图谱如图1a和1b所示。图1a的N1s XPS谱图中结合能为398.2 eV和 399.8 eV的峰分别归属于以吡啶N和吡咯N形式存在的氮元素;而图1b所示C1s XPS谱图中284.6 eV的尖锐峰归属为石墨烯中sp2杂化状态的C,288.0 eV处的宽峰为GO中处于氧化状态的C (-C=O),而在285.8 eV出现的峰归属为与N原子结合的C原子;可能由于掺入的P元素少,在该样品的P2p XPS图谱中并未发现明显的峰。
同样采用共掺杂方法但增加了P/N比例所获得的N1P1.7-GO样品,其P2p XPS谱图中明显观测到了N-P键(如图1c插图所示)[23-24];此外,在其N1s XPS谱图(图1c)中出现了三种化学环境的N元素,除吡咯N与吡啶N外的401.4 eV处的峰归属于石墨N;而O1s XPS(图1d)中可以看到部分N与O形成了N-O键。对比N1P0.37-GO和N1P1.7-GO XPS表征结果可知,在氮磷共掺杂的过程中较少量P元素有助于N元素以吡咯N的形式掺入石墨烯碳骨架中;而若P元素过多则会导致在N和P原子在石墨烯碳骨架的边缘或缺陷处与C原子结合,且相邻的N和P原子之间形成了类键合的相互作用(结构示意图如图2a所示)。
采用顺序掺杂获得的N1-P1.7-GO样品XPS谱图如图1e-1f所示。值得注意的是,未发现归属为吡咯N与吡啶N的峰,而在结合能为402.28 eV与403.99 eV处出现了N-O与O-N-O特征峰,同时在P2p XPS谱图(图1e插图)显示P以N-O-P和P-OH的形式存在,此外O1s XPS谱图(图1f)也印证了N-O与P-O的存在。综上判定在N1-P1.7-GO样品中P元素以磷酸酯的形式与N相连(结构示意图如图2b所示),推测N改性GO与磷酸共混时,磷酸优先与骨架中较活泼的N原子形成磷酸酯键,而不是碳原子。这种磷酸酯结构的形成有可能封闭了吡咯N与吡啶N作为氧还原活性位点的功能,从而影响改性GO的2e-ORR性能。

图1 XPS谱图:(a) N1P0.37-GO的N1s; (b) N1P0.37-GO 的C1s;(c) N1P1.7-GO的N1s和P2p; (d) N1P1.7-GO的O1s; (e) N1-P1.7-GO的N1s和P2p; (f) N1-P1.7-GO的O1s

图2 催化剂中N、P元素掺杂方式示意图:(a) N1P1.7-GO;(b) N1-P1.7-GO
2.2 改性GO的ORR催化性能表征结果
如图3a所示为氧气气氛下负载有N1P0.37-GO的电极在2 mol/L NaOH溶液中的CV曲线,在0.63 V处出现的还原峰归因于氧气在阴极上的还原过程。RRDE装置上不同转速ω下的极化曲线如图3c所示,取扩散控制区半波电位0.63 V的电流密度J,以1/J对ω-1/2作图进行线性拟合,根据Koutechy-Levich方程计算,得到转移电子数n=2.3;且在0.2~0.6 V范围内2e-ORR选择性均达到了85%以上,最高约达89%,表明N1P0.37-GO表现良好的2e-ORR催化性能。相同表征方法结果表明负载N1P1.7-GO的电极的氧还原过程转移电子数n=2.6,意味着N1P1.7-GO的2e-ORR活性略低于N1P0.37-GO。
2.3 改性GO催化氧还原制备H2O2的性能
以负载有NP改性GO的电极为阴极搭建电解槽进行电解实验,基于电解液中H2O2浓度评价其催化氧还原制备H2O2的性能,并与未掺杂GO样品进行比较,结果如图4所示。可以看出,未掺杂GO自身也可以表现较好的2e-ORR性能,电流效率达到59.92%;氮磷共掺杂得到的N1P0.37-GO和N1P1.7-GO样品制备H2O2的电流效率均比未掺杂GO有所提高,分别为93.27%和76.64%;然而,氮磷顺序掺杂得到的N1-P1.7-GO样品仅表现出31.20%的电流效率。
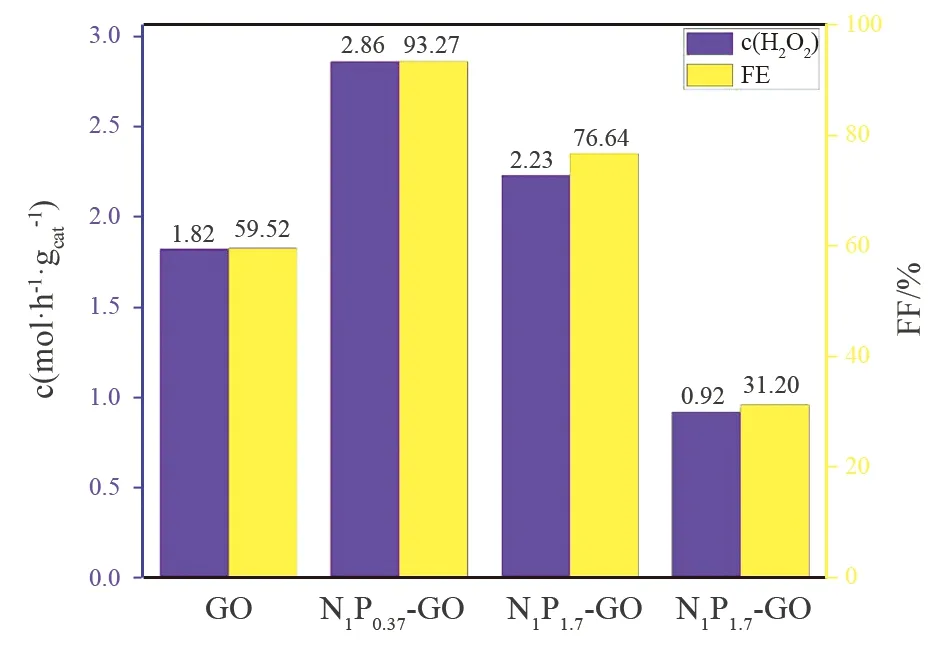
图4 GO及改性GO催化氧气还原制备H2O2的效果
上述结果证实,N、P元素在石墨烯碳骨架中的不同存在状态影响氧气分子在电极表面的反应过程,从而影响碳材料的2e-ORR催化性能。由前述结构表征结果可知,在共掺杂法改性GO制备过程中少量磷酸的共存促进了N原子主要以吡咯N形式进入碳骨架中,据文献报道这种形式的N原子存在有助于碳材料对于OOH*中间态具有合适的吸附能力,使得N1P0.37-GO表现良好的2e-ORR性能。当共掺杂过程中加入过量磷酸时,增加了P原子与N原子的相邻几率,而相邻的N与P原子间的弱相互作用限制了N原子作为氧还原活性位点的功能,导致N1P1.7-GO的2e-ORR催化性能相比于N1P0.37-GO有所降低。另一方面,在得到了N改性GO后再以磷酸进行P掺杂时,磷酸倾向于与碳骨架中的N原子形成N-O-P的磷酸酯结构。这种磷酸酯结构首先构成了很大的位阻,阻碍了氧气分子在电极表面的吸附过程,其次封闭了N原子的氧还原活性位点功能,最终导致N1-P1.7-GO制备过氧化氢的电流效率甚至低于未掺杂GO。
3 结论
以磷酸为磷源、尿素为氮源采用水热法对氧化石墨烯(GO)进行改性,分别采用共掺杂和顺序掺杂方式和调整氮磷投料比方法制备了三种氮磷改性GO。综合结构表征和电化学催化性能结果可知,在共掺杂过程中少量磷酸的存在有助于N元素以吡咯氮形式进入碳骨架,从而表现良好的2e-ORR催化性能;而如果磷酸投料量过多,则会造成碳骨架中N和P原子相邻形成弱相互作用的概率增加,从而限制了吡咯氮作为氧还原活性位点的功能,使得电流效率降低;而若先掺杂氮再掺杂磷,则磷酸倾向于与骨架中的N原子而不是C原子发生反应,形成的磷酸酯结构不但限制了氧气分子的吸附过程也阻碍了OOH*中间体的吸脱附,导致2e-ORR效率显著降低。本研究系统研究了N和P原子在碳骨架中的存在形式与其2e-ORR催化性能之间的构效关系,为后续高性能2e-ORR催化剂的研发奠定了基础。
猜你喜欢
四川轻化工大学学报(自然科学版)(2021年3期)2021-08-30 06:36:44
湿法冶金(2020年1期)2020-02-24 06:22:04
科学与财富(2017年9期)2017-06-09 18:45:34
中国氯碱(2017年3期)2017-04-18 02:23:04
电镀与环保(2016年3期)2017-01-20 08:15:29
合成化学(2015年10期)2016-01-17 08:56:06
发明与创新(2015年37期)2015-02-27 10:40:35
华东师范大学学报(自然科学版)(2014年4期)2014-03-11 16:18:28
无机化学学报(2014年4期)2014-02-28 17:31:16
机电信息(2014年5期)2014-02-27 15:51:48
