
头花蓼提取物对左氧氟沙星血浆蛋白结合率的影响
2022-09-13 03:57王玲袁丽郑林巩仔鹏李月婷潘洁黄勇贵州医科大学贵州省药物制剂重点实验室药用植物功效与利用国家重点实验室贵阳550004贵州医科大学药学院贵阳550004贵州医科大学民族药与中药开发应用教育部工程研究中心贵阳550004
中南药学 2022年8期
王玲,袁丽,郑林,巩仔鹏,李月婷,潘洁,黄勇*(.贵州医科大学贵州省药物制剂重点实验室/药用植物功效与利用国家重点实验室,贵阳 550004;2.贵州医科大学药学院,贵阳 550004;.贵州医科大学民族药与中药开发应用教育部工程研究中心,贵阳 550004)
左氧氟沙星作为喹诺酮类广谱抗菌药物,在临床上广泛用于治疗泌尿生殖系统感染、呼吸道感染和皮肤软组织感染等[1],在治疗尿路感染方面疗效尤为显著。头花蓼(Polygonum capitatum)是一种贵州苗族习用药材,具有清热利湿、利尿通淋、解毒止痛等功效[2]。临床上头花蓼提取物制成的单方制剂热淋清颗粒与左氧氟沙星联用十分广泛,联合使用可提高疗效,缩短疗程,减少抗菌药物的不良反应[3-5]。尽管两者联合应用的临床疗效评价研究较多,但尚未见国内外文献有关于头花蓼提取物对左氧氟沙星血浆蛋白结合率影响方面的研究报道。药物血浆蛋白结合率是药代动力学的重要参数之一,血浆蛋白结合率的变化可引起药物血药浓度的变化,从而改变其药代动力学行为,在临床联合用药方面有重要意义[6]。
中药的成分复杂,绝大多数情况是吸收入血的成分才可以发挥其疗效,其中各成分不仅与化学药产生相互作用,成分间也会相互竞争蛋白结合位点。因此,本文采用连续灌胃大鼠头花蓼提取物后的含药血浆与左氧氟沙星,同时,基于2×2析因设计,以左氧氟沙星的浓度和作用方式为考察因素,研究两个因素的主效应以及其交互作用[7],可更真实客观地了解两药联用后在体内与血浆蛋白结合的过程。本文将采用平衡透析法结合UPLC-MS/MS法对左氧氟沙星与大鼠血浆蛋白的结合进行测定,探讨头花蓼提取物对左氧氟沙星蛋白结合率的总体影响,为进一步研究两者在临床联合用药的安全合理性及药代动力学特征提供实验依据。
1 材料
1.1 仪器
ACQUITY UPLC超高效液相色谱仪,TQ-D三重四极杆质谱仪(包括自动进样器、二元梯度泵、柱温箱、真空脱气机、Masslynx 4.1质谱工作站,美国沃特世公司);高速离心机(Allegra X-30 R,美国贝克曼公司);超声波清洗器(KQ-300DE,昆山市超声仪器有限公司);氮气吹干仪装置(MTN-2800D,天津奥特塞恩斯仪器有限公司);多管涡旋振荡器(VX-Ⅲ,北京踏锦科技有限公司);万分之一电子天平(EL204,上海METTLER TOLEDO公司);实验室专用超纯水机(WP-UP-III-20,四川沃特尔科技发展有限公司);微量移液器(德国eppendorf股份公司);低温冰箱(BD-328WL,海尔集团公司);透析袋MD-25kD(截留相对分子量8000~14 000,塞兰博生物制品有限公司)。
1.2 试药
头花蓼提取物[参照《中国药典》[8]热淋清颗粒的提取方式提取,出膏率:9.32%,没食子酸含量:42.06 mg·g-1)];左氧氟沙星对照品(批号:130455-201607,纯度:97.3%)、葛根素对照品(批号:110752-201512,纯度:95.4%)(中国食品药品检定研究院);甲酸、甲醇(色谱纯,德国默克公司);纯净水(广州屈臣氏食品饮料有限公司);其他试剂均为分析纯。
1.3 动物
SPF级SD大鼠,雌雄各半,体质量为(230±10)g [长沙天勤生物技术有限公司,许可证号:SCXK(湘)2019-0014]。饲养条件:动物房照明充足(12/12 h光/暗周期调节),空调通风良好,相对湿度(50±10)%,室温(22±2)℃,定期对实验室进行消毒。
2 方法
2.1 溶液的配制
2.1.1 对照品溶液和内标的配制
① 精密称取左氧氟沙星适量,用甲醇定容至10 mL,即得左氧氟沙星对照品(1.002 mg·mL-1)的储备液,置-20℃保存,备用。
② 精密称取葛根素(4.722 mg),用50%甲醇定容至10 mL,即得葛根素(0.4722 mg·mL-1)的储备液。取内标葛根素储备液适量至100 mL量瓶中,用50%甲醇定容至刻度,配制成1µg·mL-1的内标溶液,置-20℃保存,备用。
2.1.2 磷酸缓冲液(PBS)的配制 精密称取KH2PO40.2 g,Na2HPO42.88 g,KCl 0.2 g,NaCl 8.0 g,用适量超纯水溶解,加1% NaOH调节pH 7.4,将溶液转移至1000 mL量瓶中,超纯水定容至刻度,即得0.01 mol·L-1PBS溶液,储存备用。
2.2 色谱条件
色谱柱:Waters BEH C18(2.1 mm×100 mm,1.7µm)柱;保护柱:Waters Van Guard BEH C18(2.1 mm×5 mm,1.7 µm)柱;柱温:45℃,进样器温度:25℃;流速:0.30 mL·min-1,进样体积:1µL;流动相:0.1%甲酸乙腈(A)-0.1%甲酸水(B),梯度洗脱(0~0.3 min,10%A;0.3~1.5 min,10%~40%A;1.5~3 min,40%~90%A;3~3.2 min,90%A;3.2~4.0 min,90%~10%A)。
2.3 质谱条件
采用电喷雾电离源(ESI);正离子模式扫描;毛细管电离电压:3 kV;离子源温度:120℃;喷雾气与反吹气:N2;去溶剂气流速:650 L·h-1,去溶剂气温度:350℃;扫描方式为多反应离子监测(MRM),质谱数据采集及处理软件为Masslynx 4.1质谱工作站。左氧氟沙星及内标用于定量分析的监测离子见表1。

表1 左氧氟沙星及内标的质谱条件Tab 1 Mass spectrometry conditions for the levofloxacin and internal standards
2.4 样品处理方法
精密量取血浆样品(透析内液)和缓冲液(透析外液)各100 µL,置1.5 mL EP管中,加入2%甲酸水50 µL,涡旋混匀(1 min),加入50 µL葛根素内标溶液(1 µg·mL-1),再加入甲醇400 µL,涡旋混匀(1 min),超声10 min,离心(12 000 r·min-1、4℃,10 min),取上清液置EP管中,37℃ N2吹干,150 µL 50%甲醇复溶,涡旋混匀1 min,超声10 min,离心(12 000 r·min-1、4℃,10 min),取上清液进样分析。
2.5 统计学分析
实验数据采用SPSS 22.0统计软件进行分析,结果用±s表示,组间比较采用单因素方差分析,各因素间交互作用采用析因设计方差分析,P<0.05为差异有统计学意义。
2.6 方法学考察
2.6.1 专属性 取100 µL大鼠空白血浆(不加内标)、加入对照品和葛根素的血浆、大鼠给药后的血浆,按“2.4”项下方法操作,进样记录各血浆样品色谱图,考察方法专属性。
2.6.2 标准曲线的制备和LLOQ 取左氧氟沙星对照品储备液适量,分别加入空白血浆和空白缓冲液,得到在血浆中质量浓度为1.002、2.505、5.010、10.020、20.040、25.050、30.060 μg·mL-1的对照品溶液,在缓冲液中的质量浓 度 为1.002、2.505、5.010、10.020、20.040、25.050 μg·mL-1的对照品溶液。按“2.4”项下方法操作,以待测成分与相应葛根素内标峰面积之比(A/Ai)为纵坐标y,各物质浓度(C)为横坐标x进行线性回归,即得标准曲线。左氧氟沙星的定量下限(LLOQ)定义为S/N=10。
2.6.3 准确度和精密度 分别配制左氧氟沙星大鼠血浆低、中、高(2.505、10.020、25.050 μg·mL-1)和缓冲液低、中、高(2.505、10.020、20.040 μg·mL-1)三种质量浓度的质控(QC)样品,各浓度平行5份,按“2.4”项下方法处理,日内连续进样,连续测定3 d,计算日内和日间精密度。同法平行配制低、中、高三个浓度的QC样品5份,将测得峰面积代入随行标准曲线计算左氧氟沙星的实际浓度,以实际测得左氧氟沙星的浓度与加入时左氧氟沙星浓度之比计算准确度。
2.6.4 提取回收率和基质效应 取100 µL大鼠空白血浆和缓冲液,按“2.6.3”项下方法分别配制低、中、高三个浓度的QC样品,每种浓度平行配制5份,按“2.4”项下操作(A样品);另取100 μL空白血浆和缓冲液,除不加左氧氟沙星对照品溶液外,其他按“2.4”项下方法操作,向离心后获得的上清液中加入相应低、中、高三种浓度的左氧氟沙星对照品溶液和内标葛根素,37℃N2吹干,残留物以50%甲醇150 μL溶解(B样品);另取上述低、中、高三种浓度的左氧氟沙星对照品溶液与内标葛根素,37℃ N2吹干,残留物以50%甲醇150 μL溶解(C样品)。以A样品与B样品得到的色谱峰面积之比计算提取回收率,以B样品与C样品得到的色谱峰面积之比计算基质效应。
2.6.5 样品稳定性 按“2.6.3”项下方法配制左氧氟沙星的空白血浆和缓冲液低、中、高三个浓度QC样品,考察样品处理后分别置于室温6 h(约20℃)中,4℃下冷藏12 h,反复冻融3次三种条件下的稳定性。重复5样本分析。
2.7 析因实验设计
采用2×2析因实验设计,A因素(浓度)和B因素(作用方式)各选择2个水平:A1(3 μg·mL-1)、A2(6 μg·mL-1),B1(左氧氟沙星)、B2(头花蓼提取物预处理),共4个组,每组重复6次,实验方案见表2。
表2 2×2析因设计及实验方案Tab 2 2×2 factor design and experiment plan
2.8 血浆蛋白结合率测定
将大鼠分为两组:左氧氟沙星组(股动脉取空白血)及头花蓼提取物预处理组[连续给予大鼠头花蓼提取物(1.86 g·kg-1,参照前期药代动力学实验研究[9]的剂量)3 d],一天两次,末次给药2 h后股动脉取血。
参照文献[10],将透析袋进行预处理,处理后的透析袋一端折叠用线结扎。按照析因实验分组,取1 mL空白血浆(左氧氟沙星组)或取1 mL灌胃头花蓼提取物的血浆(头花蓼提取物预处理组)加入透析袋内,透析袋内留一小空气泡,将透析袋另一端扎紧,使其悬浮于盛有20 mL缓冲液的广口瓶中,膜外缓冲液左氧氟沙星质量浓度分别为3 μg·mL-1、6 μg·mL-1,调节透析袋内外液面,使其保持在同一水平面,并避免透析袋贴于瓶壁。将瓶口密封后,于4℃静置48 h至药物扩散平衡。透析结束后,用10%的高氯酸溶液检查透析外液是否有蛋白漏出,有漏出者则该样品作废。分别测定透析袋内药物浓度Dt(总浓度)与袋外药物浓度Df(游离药物浓度),根据公式计算血浆蛋白结合率:血浆蛋白结合率(%)=(Dt-Df)/Dt×100%。
3 结果
3.1 方法学考察结果
3.1.1 专属性 空白血浆、空白血浆+左氧氟沙星对照品+内标葛根素、大鼠给药后血浆色谱图见图1,表明血浆中内源性物质不干扰各个成分的测定。
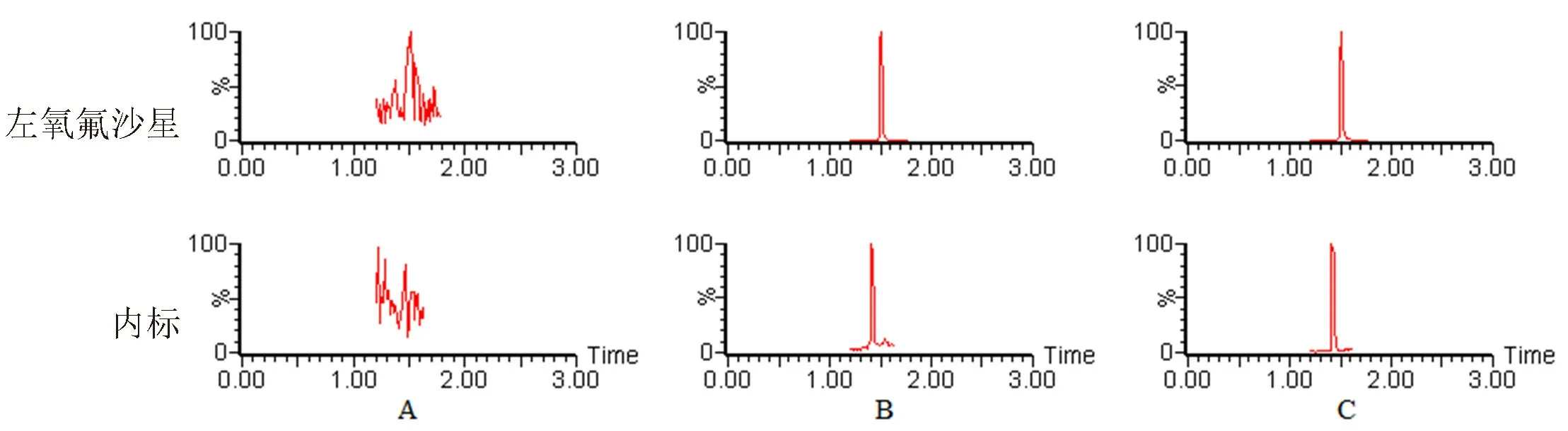
图1 典型UPLC-MS/MS色谱图Fig 1 Typical UPLC-MS/MS chromatograms
3.1.2 线性关系及定量下限 大鼠血浆和缓冲液中左氧氟沙星在相应的质量浓度范围内与峰面积呈良好的线性关系(见表3)。

表3 血浆和缓冲液中左氧氟沙星的标准曲线和线性范围Tab 3 Standard curve and linearity of levofloxacin in plasma and buffer
3.1.3 准确度和精密度 左氧氟沙星在大鼠血浆和缓冲液中的日内精密度和日间精密度RSD均小于15%,准确度为89.12%~ 106.16%,提示该方法准确、可靠、重现性好,符合生物样品分析方法要求(见表4)。
表4 左氧氟沙星在大鼠血浆及缓冲液中的准确度、日内和日间精密度(±s,n=5)Tab 4 Accuracy of levofloxacin in the rat plasma and the buffer,intra-day and inter-day precision (±s,n=5)
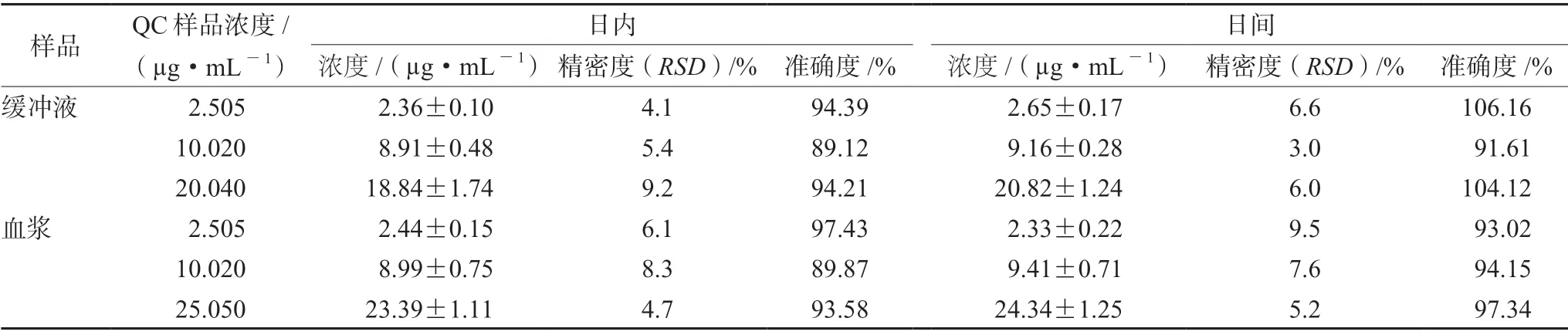
表4 左氧氟沙星在大鼠血浆及缓冲液中的准确度、日内和日间精密度(±s,n=5)Tab 4 Accuracy of levofloxacin in the rat plasma and the buffer,intra-day and inter-day precision (±s,n=5)
样品 QC样品浓度/(µg·mL-1)日内日间浓度/(µg·mL-1)精密度(RSD)/% 准确度/% 浓度/(µg·mL-1) 精密度(RSD)/% 准确度/%缓冲液 2.505 2.36±0.10 4.1 94.39 2.65±0.17 6.6 106.16 10.020 8.91±0.48 5.4 89.12 9.16±0.28 3.0 91.61 20.040 18.84±1.74 9.2 94.21 20.82±1.24 6.0 104.12血浆 2.505 2.44±0.15 6.1 97.43 2.33±0.22 9.5 93.02 10.020 8.99±0.75 8.3 89.87 9.41±0.71 7.6 94.15 25.050 23.39±1.11 4.7 93.58 24.34±1.25 5.2 97.34
3.1.4 提取回收率和基质效应 低、中、高三个浓度下左氧氟沙星提取回收率在91.11%~105.42%,基质效应在87.99%~ 103.97%,RSD均小于15%。结果表明该方法提取回收率良好,不存在明显的基质效应,均满足生物样品分析方法要求(见表5)。
表5 左氧氟沙星在大鼠血浆及缓冲液中的提取回收率和基质效应(±s,n=5)Tab 5 Extraction recovery and matrix effect of levofloxacin in the rat plasma and the buffer (±s,n=5)
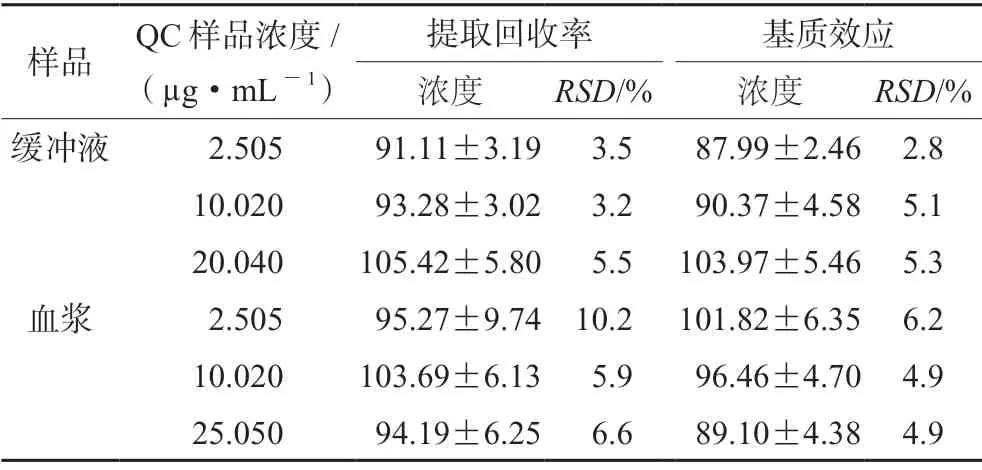
表5 左氧氟沙星在大鼠血浆及缓冲液中的提取回收率和基质效应(±s,n=5)Tab 5 Extraction recovery and matrix effect of levofloxacin in the rat plasma and the buffer (±s,n=5)
样品 QC样品浓度/(µg·mL-1)提取回收率 基质效应浓度 RSD/% 浓度 RSD/%缓冲液 2.505 91.11±3.19 3.5 87.99±2.46 2.8 10.020 93.28±3.02 3.2 90.37±4.58 5.1 20.040 105.42±5.80 5.5 103.97±5.46 5.3血浆 2.505 95.27±9.74 10.2 101.82±6.35 6.2 10.020 103.69±6.13 5.9 96.46±4.70 4.9 25.050 94.19±6.25 6.6 89.10±4.38 4.9
3.1.5 样品稳定性 左氧氟沙星血浆和缓冲液样品在室温6 h、冷藏12 h和反复冻融3次均比较稳定,满足生物样品分析方法要求(见表6)。
表6 左氧氟沙星在大鼠血浆和缓冲液中的稳定性(±s,n=5)Tab 6 Stability of levofloxacin in the rat plasma and the buffer (±s,n=5)
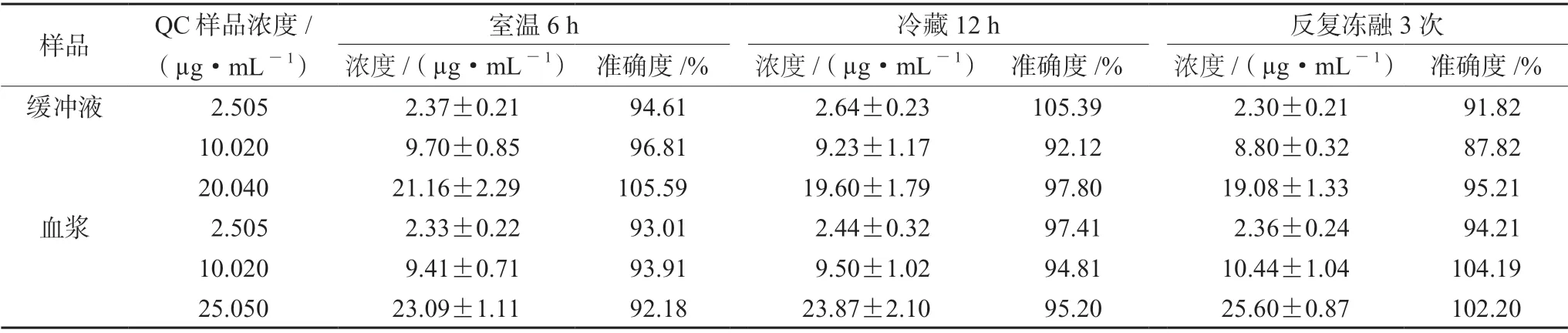
表6 左氧氟沙星在大鼠血浆和缓冲液中的稳定性(±s,n=5)Tab 6 Stability of levofloxacin in the rat plasma and the buffer (±s,n=5)
?次样品 QC样品浓度/(µg·mL-1)室温6 h 冷藏12 h 反复冻融3浓度/(µg·mL-1) 准确度/% 浓度/(µg·mL-1) 准确度/% 浓度/(µg·mL-1) 准确度/%缓冲液 2.505 2.37±0.21 94.61 2.64±0.23 105.39 2.30±0.21 91.82 10.020 9.70±0.85 96.81 9.23±1.17 92.12 8.80±0.32 87.82 20.040 21.16±2.29 105.59 19.60±1.79 97.80 19.08±1.33 95.21血浆 2.505 2.33±0.22 93.01 2.44±0.32 97.41 2.36±0.24 94.21 10.020 9.41±0.71 93.91 9.50±1.02 94.81 10.44±1.04 104.19 25.050 23.09±1.11 92.18 23.87±2.10 95.20 25.60±0.87 102.20
3.2 头花蓼提取物对左氧氟沙星血浆蛋白结合率的影响
左氧氟沙星在3 μg·mL-1、6 μg·mL-1下,灌胃头花蓼提取物后,与单用左氧氟沙星组相比,联用组左氧氟沙星的血浆蛋白结合率均显著增加(P<0.05)。随着左氧氟沙星浓度增加,其蛋白结合率均降低(见表7)。析因方差分析结果见表8,各因素对左氧氟沙星的蛋白结合率的影响大小为:FB>FA>F(A*B),表明作用方式因素对左氧氟沙星的蛋白结合率影响大于浓度因素。
表7 头花蓼提取物对左氧氟沙星血浆蛋白结合率的影响(±s,n=6)Tab 7 Effect of Polygonum capitatum extract on the plasma protein binding rate of levofloxacin (±s,n=6)

表7 头花蓼提取物对左氧氟沙星血浆蛋白结合率的影响(±s,n=6)Tab 7 Effect of Polygonum capitatum extract on the plasma protein binding rate of levofloxacin (±s,n=6)
注(Note):与单用组相比,*P<0.05(Compared with the single group,*P<0.05)。
?

表8 主效应及交互作用分析结果Tab 8 Main effect and interaction analysis
4 讨论
目前,已报道的关于研究药物血浆蛋白结合率的方法,主要包括平衡透析法、超滤法、超速离心法、凝胶过滤法等[11],其中平衡透析法具有操作简单、经济、受实验因素干扰小等优点[12],故采用最为经典的方法来测定血浆蛋白结合率。据文献报道,血浆蛋白分子量为69 kD,考虑到血浆中大量内源性物质透析到袋外,可能对血浆蛋白的结合能力有影响,因此本实验采用截留分子量为 8000~14 000的25 kD透析袋进行实验,效果良好[13]。利用平衡透析法测定蛋白结合率最经典的方法是在透析袋内加入空白血浆,外液放入含药PBS溶液,而本文首次将头花蓼提取物多次灌胃后取其血浆研究对左氧氟沙星蛋白结合率的影响。中药具有“多成分、多环节、多途径、多靶点”的作用特点,因此,灌胃后取其血浆更能真实地反映头花蓼口服进入胃肠道后吸收入血的过程,再用体外实验进行蛋白结合率的研究,更能客观反映两药合用后其在体内的蛋白结合情况。
本实验采用UPLC-MS/MS技术结合平衡透析法测定左氧氟沙星的蛋白结合率,结果表明该法可快速、准确地测定左氧氟沙星的含量。本研究采用平衡透析法,对实验平衡时间进行了考察,在4℃条件下,平衡12、24、36、48和72 h后分别测定透析袋内外浓度,结果在平衡48 h后,左氧氟沙星与血浆蛋白结合已达到平衡,最终选择48 h为平衡时间。同时,本课题组前期药代动力学实验表明,大鼠给予左氧氟沙星后Cmax为(6032.91±1960.11)ng·mL-1,因此,选择6 μg·mL-1和3 μg·mL-1两个浓度的左氧氟沙星进行头花蓼提取物对左氧氟沙星蛋白结合率的影响研究[9]。析因方差分析结果表明,两者之间不存在明显的交互作用。其中浓度和作用方式对左氧氟沙星的蛋白结合率均有显著影响,但尚不能认为是两者间的交互作用对左氧氟沙星的蛋白结合率产生影响。
左氧氟沙星的血浆蛋白结合率约为20%~22%,与文献报道血浆蛋白结合率24%~38%存在差异[14],可能与透析膜的微量吸附有一定关系。另外,左氧氟沙星与大鼠血浆蛋白低度结合,说明其由于血浆蛋白结合率的改变而导致游离药物浓度急剧升高所产生不良反应的可能性不大,因此,增加了与其他药物联用的可能性。药物与血浆蛋白结合的变化影响血液中游离药物浓度,导致药物在体内的分布、代谢、排泄以及目标靶点结合的变化,从而影响药理效应过程。本课题组前期在组织分布与排泄实验中,头花蓼提取物和左氧氟沙星合用后左氧氟沙星在各组织中含量显著降低,尿累积排泄率从20.69%下降到11.84%,粪便累积排泄率从26.08%下降到13.28%[15]。本研究结果表明联合应用头花蓼提取物后左氧氟沙星的蛋白结合率显著增加(P<0.05),药物从血液进入各组织和器官中的药量减少,影响药物在体内的分布和排泄过程,这与前期实验研究结果基本一致。临床研究报道将头花蓼提取物与左氧氟沙星联用来提高其治疗泌尿系统感染的疗效,目前,在药代动力学水平上的研究无法强有力地解释这个问题。药物相互作用可分为药代动力学相互作用和药效学相互作用。在药效学层面,药物如何使机体发挥作用,其作用机制是全面而复杂的,推测可能是由于两种药物对病原菌的协同抑制作用[16-17],降低细菌在尿路上皮细胞中的黏附能力,加快致病菌排出体外,从而提高疗效,但是这些推测还需要进一步的实验来证实。因此,通过头花蓼提取物对左氧氟沙星血浆蛋白结合率影响的研究,可为两者临床合理用药体内过程研究提供更全面的药代动力学参数,后期可对两者合用的具体机制进行更深入的研究。
猜你喜欢
科技视界(2022年10期)2022-05-20
中国典型病例大全(2022年12期)2022-05-13
大众健康(2022年4期)2022-04-27
康颐(2020年15期)2020-11-10
昆明医科大学报(2019年2期)2019-09-10
新教育时代·教师版(2016年6期)2016-05-14
中国民族民间医药·下半月(2014年2期)2014-09-26
中国新闻周刊(2014年32期)2014-05-14
