
K-Ni-Mo基催化剂的水热还原法制备及用于合成气制低碳醇反应性能研究
2022-09-09 02:23孙付琳王乾浩房克功汪颖军
燃料化学学报 2022年8期
孙付琳 ,赵 璐 ,王乾浩 ,房克功 ,汪颖军
(1.东北石油大学 化学化工学院, 黑龙江 大庆 163318;2.中国科学院山西煤炭化学研究所 煤转化国家重点实验室, 山西 太原 030001)
中国煤炭资源丰富,实施“以煤代油”的战略措施可替代和补充油气资源,也关系到中国经济与能源领域的安全稳定。其中,合成气制低碳醇(含2个及2个以上碳原子的醇)是非石油路径制取液态清洁燃料、油品添加剂和大宗化学品的重要技术方法,也是碳一化学研究的重要组成部分[1,2]。在合成气制低碳醇技术中,高效催化剂的设计、制备及其反应性能的研究作为推动该项技术的基石已受到研究者广泛关注。
目前,合成气制低碳醇催化剂主要分为以下几类:改性甲醇催化剂[3-5]、改性费托催化剂[6-11]、Rh基催化剂[12,13]和Mo基催化剂[14,15]等。改性甲醇催化剂的产物主要以醇类为主,但甲醇选择性高,催化剂易烧结失活。改性费托催化剂反应条件温和,低温CO转化率高,但该类催化剂热稳定性差,产物中烃类选择性较高。Rh基催化剂具有高乙醇选择性,但贵金属Rh价格昂贵,限制了其在工业生产中的大规模应用。以MoS2为代表的Mo基催化剂因其优异的抗硫性能,延长了反应周期,并且具有相对高的醇类选择性,但反应条件相比金属催化剂苛刻,产物中会引入含硫杂质。如何开发新型高效非硫化态钼基催化剂,在避免引入杂质硫的同时又能在温和条件下有效提高反应活性和低碳醇选择性,是当前针对钼基催化剂研究的重难点[15,16]。
在低碳醇合成用钼基催化剂的研究中采用碱金属(如Na[17]、K[18]、Cs[19]等)、过渡金属(费托组元Fe[20]、Co[21]、Ni[22]等)等作为助剂对其进行改性,从而有助于提高低碳醇选择性。相较其他碱金属,K作为助剂表现出更优的促进低碳醇生成性能[23,24]。Liakakou等[25]发现,K助剂可改善活性相分散度,同时抑制烃类生成。Xie等[26]报道了K-CoMo/C催化剂。在钼基催化剂上添加K后,生成甲醇受到明显抑制,乙醇成为主要产物,提高了C2+OH选择性。他们认为上述结果主要归因于引入K能促进烷基(CnHx)的形成,而CnHx被认为是合成低碳醇的关键中间体。而针对过渡金属助剂的研究发现,加入Ni在促进C–C键耦合及碳链增长的同时,对H2仍具有良好的吸附活化性能[27]。Ma等[28]在考察Ni含量对Mo基催化剂的影响时,发现Mo基催化剂表面高浓度的活性H物种有助于抑制副反应,同时催化剂表面Ni物种的百分含量与低碳醇选择性呈现正相关趋势。已有研究显示以低碳醇为目标产物,催化剂需同时具有催化碳链增长和含氧化功能的紧密接触活性位,而钼基催化剂的催化活性和醇选择性主要取决于助剂与钼基活性相的紧密接触[29]。这是由于助剂与钼基活性相的紧密接触有利于两者间的电子转移和吸附物种的迁移[30]。然而,多数报道集中在以K2CO3与费托改性钼基活性相通过机械混合制备[31,32],并且由于费托组元极易与Mo物种形成金属钼酸盐物相,从而导致催化剂低温加氢活性偏低[25,28,33]。
本研究为解决上述问题,采用水热合成法首先制备出Ni-Mo基催化剂前体,随后再引入K经还原处理制得具有金属Ni和K2MoO4紧密接触的非硫化态K-Ni-Mo基催化剂,并考察了其催化合成气制低碳醇反应性能。利用XRD、N2物理吸附-脱附、H2-TPR、HR-TEM、SEM-EDS、XPS、H2-TPD、CO-TPD和CO2-TPD等手段深入探究了所合成不同K含量的非硫化态K-Ni-Mo基催化剂其化学和物理性质与合成气制低碳醇反应性能之间的构效关联。
1 实验部分
1.1 催化剂的制备
首先采用水热法合成实验用Ni-Mo基催化剂前体。所有试剂原料均为化学分析纯。称取3.0 g葡萄糖溶于20 mL蒸馏水中得到溶液A,将1.8 g(NH4)6Mo7O24·4H2O和11.6 g Ni(NO3)2·6H2O溶 于30 mL蒸馏水并搅拌30 min得到溶液B,在混合溶液A和B后转入聚四氟乙烯反应釜,置于180 ℃下保持48 h。随后经离心、洗涤处理得到固体样品,在80 ℃真空干燥箱中陈化12 h,最后于氮气气氛下400 ℃焙烧4 h,得到Ni-Mo基催化剂前体。将一定量K2CO3与Ni-Mo基催化剂混合,后经400 ℃焙烧、330 ℃还原处理得到具有不同K含量的非硫化态Kx-Ni-Mo基催化剂(x=0、0.1、0.2、0.3、0.4、0.5,x表示K/(Ni+Mo))。
1.2 催化剂的表征
采用D8 Advance Bruker AXS衍射仪进行不同催化剂的XRD表征,以CuKα为辐射源,其中,管电压为40 kV。各催化剂的织构参数和氮气吸附–脱附曲线在JW-BK200型N2吸附-脱附仪上测定。在JEOL-2100上对催化剂进行TEM分析,并在JSM-7001F上进行催化剂元素分布的SEM-EDS分析。XPS表征在AXIS ULTRADLD 型X射线光电子能谱仪上进行,采用Al靶Kα为X射线源。H2-TPR实验在常压石英管中进行,称取0.03 g(40–60目)样品,采用Ar在150 ℃下吹扫30 min,随后将气体切换至还原气氛(5% H2-95% N2、30 mL/min),以10 ℃/min升温速率升至指定温度。此外,各催化剂的H2、CO和CO2-TPD表征分析在上述相同石英管反应器中进行,具体操作步骤如下。称取0.15 g样品,在原料气H2/CO = 2、330 ℃条件下处理4 h,后在Ar下冷却至室温进行H2、CO和CO2等相应吸附实验,待催化剂表面吸附至饱和状态用Ar吹扫30 min,以10 ℃/min升温速率在Ar气氛下升至指定温度,过程中通过质谱仪检测m/z=2∶28∶44(H2/CO/CO2)信号。
1.3 CO加氢合成低碳醇反应性能评价
图1示出了本研究中所用反应装置的示意图,采用内径10 mm的不锈钢管进行CO加氢合成低碳醇性能测试。将1.5 g催化剂(40–60目)和1.5 g石英砂(40–60目)混合均匀后装于反应管中。将催化剂处理6 h(H2/CO = 2、330 ℃)。然后反应器温度冷却至30 ℃,标定进气流量。调节反应温度至240 ℃,反应压力5 MPa,空速5000 h-1进行反应评价。在反应24 h后进行取样分析,通过热阱和冷阱收集液样产物。采用气液相色谱分别对气、液产物分析。对原料气和反应后尾气采用气相色谱仪分析。H2、CO采用碳分子筛色谱柱通过TCD检测,以Ar为载气。CO、CH4、CO2采用碳分子筛色谱柱通过TCD检测,以He为载气。对尾气中的烷烃和烯烃(C1–7)采用Al2O3色谱柱通过FID检测,以Ar为载气。对产物中醇水样品使用Agilent 7890B气相色谱,通过DB-FFAP毛细管柱进行分析。
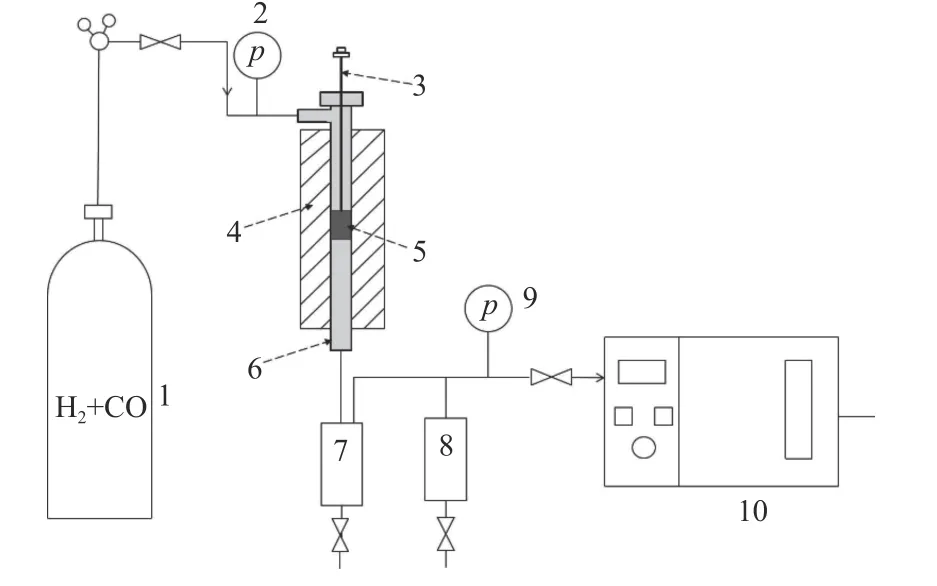
图1 合成气制低碳醇反应装置示意图Figure 1 Schematic diagram of reaction device for higher alcohols synthesis from syngas
2 结果与讨论
2.1 XRD分析
图2示出了K-Mo、Ni-Mo基及不同K含量的K-Ni-Mo基催化剂XRD谱图。Ni-Mo基催化剂和不同K-Ni-Mo基催化剂上在2θ= 37.2°、43.3°、62.9°和75.4°处出现了NiO的特征衍射峰,分别对应于NiO的(111)、(200)、(220)和(311)晶面。此外,在Ni-Mo基催化剂上还可清晰观察到对应于NiMoO4的特征衍射峰。但注意到随着K含量的增加,K-Ni-Mo基催化剂上NiMoO4衍射峰强度显著减弱,同时伴随出现K2MoO4特征衍射峰。这归因于引入K会导致K-Ni-Mo基催化剂中NiMoO4分解从而含量降低,同时伴随着K2MoO4和NiO的生成。另外,观察无Ni存在的K-Mo基催化剂XRD表征结果也发现了K2MoO4特征衍射峰,并且仅存在K2MoO4(JCPDS #29-1021)一种晶型。而值得注意的是,含Ni的K-Ni-Mo基中除了出现K2MoO4(JCPDS #29-1021)晶型外,还可观察到另一种K2MoO4(JCPDS #48-0036)晶型。已有文献报道指出,当存在杂原子时,会导致具有多种晶型的物相发生部分晶型转换。如Zhang等[34]报道纤锌矿ZnS(JCPDS #36-1450)因Cu原子存在时,会有部分晶相转变为闪锌矿ZnS(JCPDS #05-0566)。Kryshtab等[35]也报道了类似的结论。因此,出现这一现象可归因于Ni存在下导致所合成的K-Ni-Mo基催化剂中存在两种K2MoO4晶型。
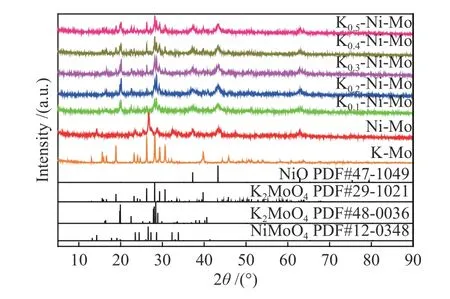
图2 K-Mo、Ni-Mo基及不同K含量的K-Ni-Mo基催化剂前体的XRD谱图Figure 2 XRD patterns of the precursors of the K-Mo, Ni-Mo and K-Ni-Mo-based catalysts with different contents of K
图3示出了Ni-Mo基以及不同K含量的K-Ni-Mo基催化剂经还原处理后的XRD谱图。如图所示,各还原后催化剂中NiO衍射峰逐渐减弱,同时在2θ= 44.5°、51.8°和76.4°处观察到Ni的特征衍射峰,分别对应于Ni的(111)、(200)和(220)晶面。各催化剂也可清晰观察到K2MoO4晶相,表明在本实验条件下K2MoO4相能稳定存在。在各不同K含量的K-Ni-Mo基催化剂中未检测到如MoO3等物相的特征衍射峰。另外,根据下文图7(b)所示不同K含量的K-Ni-Mo基催化剂上Ni 2p的XPS谱图可以发现,尽管随着K含量的增大,K物种会导致K-Ni-Mo基催化剂中NiMoO4分解并伴随产生K2MoO4,但各还原后催化剂中仍存在NiMoO4。由于K引入后NiMoO4含量下降并伴随其结晶度降低,因此,在K-Ni-Mo基催化剂体和还原处理后的XRD谱图中均未观测到相应NiMoO4的特征衍射峰。以上实验结果也与下述TEM表征结果相一致。
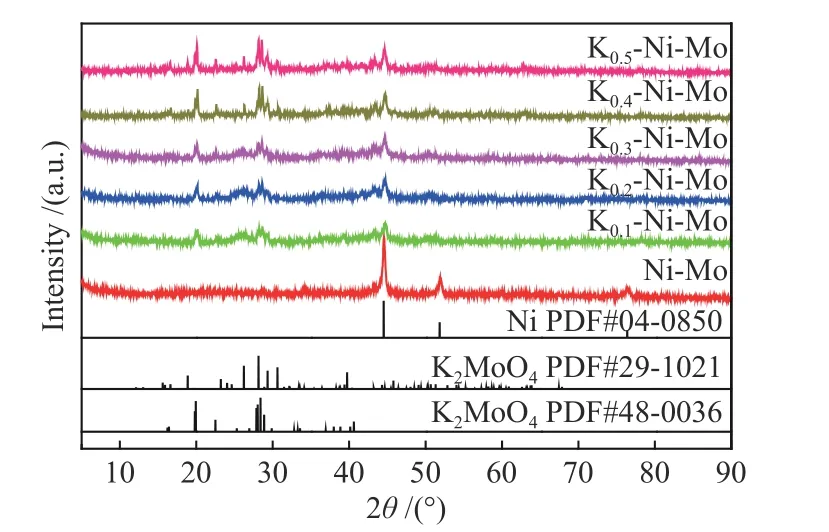
图3 经还原处理后不同K-Ni-Mo基催化剂的XRD谱图Figure 3 XRD patterns of various K-Ni-Mo-based catalysts after reduction
2.2 N2物理吸附-脱附分析
不同K-Ni-Mo基催化剂的N2吸附-脱附等温线如图4所示。各催化剂均表现出IV型等温线,具有H4型回滞环。引入不同含量K后,不同KNi-Mo基催化剂的等温线类型未发生明显变化,即未改变催化剂的孔型。表1示出了不同K含量K-Ni-Mo基催化剂的织构参数。从中可以看出未引入K的Ni-Mo基催化剂比表面积、孔容和孔径分别为72.6 m2/g、0.21 cm3/g和11.7 nm,各K-Ni-Mo基催化剂的比表面积和孔容随着K含量增加呈降低趋势,而孔径具有增大趋势。
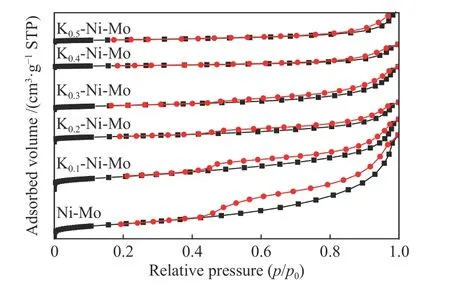
图4 不同K-Ni-Mo基催化剂的氮气吸附-脱附曲线Figure 4 Nitrogen adsorption-desorption isotherms of the KNi-Mo-based catalysts
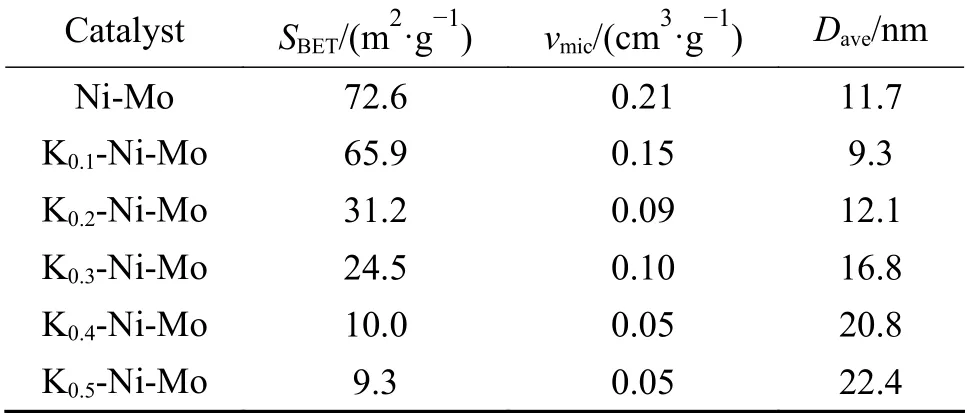
表1 不同K含量的K-Ni-Mo基催化剂的织构参数Table 1 Texture parameters of the K-Ni-Mo-based catalysts
2.3 H2-TPR分析
图5示出了K-Mo基、Ni-Mo基以及不同K含量的K-Ni-Mo基催化剂H2-TPR谱图。如图所示,K-Mo基催化剂上并未观察到明显耗氢峰。结合XRD表征结果,表明在所考察温度范围内K2MoO4物相难于还原。在Ni-Mo基和不同K含量的K-Ni-Mo基催化剂上于300–600 ℃存在低温耗氢峰。此外,在Ni-Mo基和K含量较低的K-Ni-Mo基催化剂(K0.1-Ni-Mo、K0.2-Ni-Mo)上观察到600–700 ℃存在高温耗氢峰[36]。上述两类耗氢峰分别归属于催化剂中NiO相和NiMoO4相中Ni2+→Ni0[25],这与XRD分析结果相吻合。同时,观察到随着K含量的增加,K-Ni-Mo基催化剂的低温耗氢峰从500 ℃左右逐渐向更低温移动。这一实验结果是由于K易与Mo形成K2MoO4相,从而促使NiMoO4相分解产生出更多高分散的NiO相,因此,有利于还原得到Ni金属相。该结果和XRD表征中所观察到随着K含量增加,K-Ni-Mo基催化剂上NiMoO4的特征衍射峰消失,同时伴随出现K2MoO4特征衍射峰的实验结果相印证。
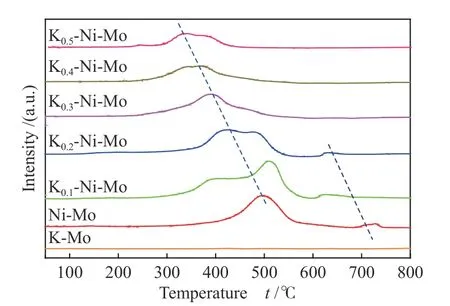
图5 不同K-Ni-Mo基催化剂的H2-TPR谱图Figure 5 H2-TPR profiles of the different K-Ni-Mo-based catalysts
2.4 HR-TEM及SEM-EDS分析
图6(a)示出了K0.4-Ni-Mo基催化剂前体的HR-TEM照片。如图所示,0.21 nm的晶格间距对应NiO(200)晶面,0.314和0.317 nm的晶格间距分别归属于K2MoO4的(130)和(310)晶面间距,这也与XRD分析结果相符。图6(b)示出了K0.4-Ni-Mo基催化剂的HR-TEM照片。其中,所观察到的0.203 nm的晶格间距与金属Ni(011)晶面的晶格间距对应。此外,TEM照片中也观察到0.314和0.317 nm的晶格间距,与氧化物前体的TEM结果相同,其分别归属于K2MoO4的(130)和(310)晶面间距。如图6(c)所示,K0.4-Ni-Mo基催化剂的SEM照片和K、Ni、Mo元素的EDS证实了K、Ni、Mo三种元素是相伴共存的,并且均匀分布,说明K0.4-Ni-Mo基催化剂在还原后保持了良好的结构稳定性。再者,也证明了引入K后形成K2MoO4相的同时促使NiMoO4相原位分解,进而伴随产生了高分散Ni物种。上述表征结果也与XRD、H2-TPR表征结果相一致。

图6 K0.4-Ni-Mo基催化剂前体的HR-TEM(a)还原后的HRTEM(b)和SEM-EDS(c)Figure 6 HR-TEM images (a) of the oxide precursors of the K0.4-Ni-Mo-based catalyst.HR-TEM (b) and SEM-EDS (c)images of the reduced K0.4-Ni-Mo-based catalyst
2.5 XPS分析
图7(a)示出了不同K含量的K-Ni-Mo基催化剂Mo 3d的XPS谱图。如图所示,在各催化剂上均可观察到Mo6+3d3/2和Mo6+3d5/2的电子结合能。对图7(a)中各XPS谱图进行分峰处理可得出分别对应于K2MoO4和MoO3中Mo6+的谱峰。观察分峰结果,对比Mo6+在K2MoO4中的电子结合能与在MoO3中的电子结合能可知,Mo6+电子结合能明显移向较低位置,以上实验结果与文献报道相符[37]。图7(b)示出了不同K含量的K-Ni-Mo基催化剂上Ni 2p的XPS谱图。根据XPS分峰结果发现,Ni元素在各K-Ni-Mo基催化剂上均出现852–856 eV的三种Ni 2p峰,分别对应于0价态金属Ni、NiO中的Ni2+和NiMoO4中的Ni2+离子,这也与上述XRD及H2-TPR分析结果相印证。
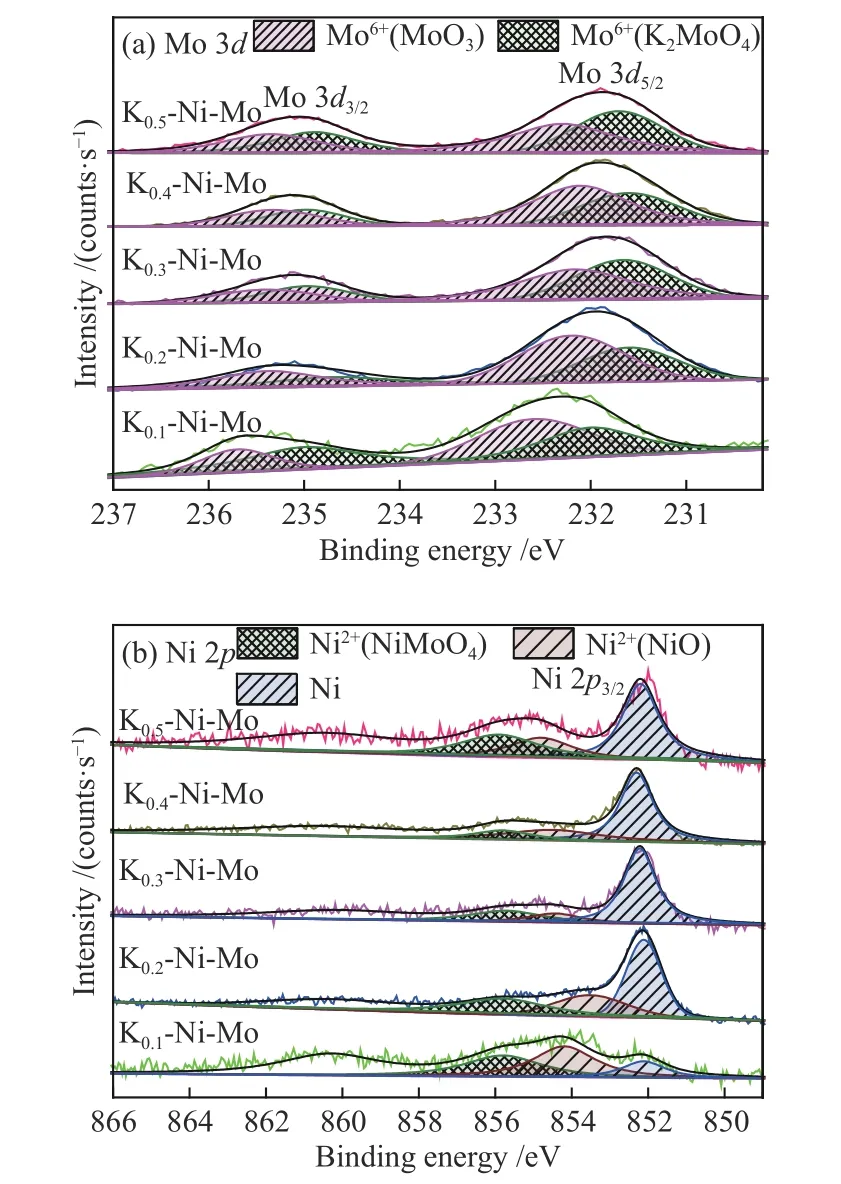
图7 不同K-Ni-Mo基催化剂的XPS谱图Figure 7 XPS spectra of the different K-Ni-Mo-based catalysts(a): Mo 3 d; (b): Ni 2 p
为了更直观地定量显示不同K含量的K-Ni-Mo基催化剂中Ni和K2MoO4含量之间关系,表2列出了通过以上XPS表征结果计算得出的不同K含量的K-Ni-Mo基催化剂中Ni/K2MoO4比。从表2中可以发现,各K-Ni-Mo基催化剂中Ni/K2MoO4比随着K含量的增加呈现先增大后降低的变化趋势。其中,在K0.4-Ni-Mo催化剂上Ni/K2MoO4比最高,约1.58。该研究结果证明K的引入可以促进Ni物种的还原,并且能够通过改变K引入量来针对性调控Ni与K2MoO4之间的相对含量。

表2 不同K含量的K-Ni-Mo基催化剂的Ni/K2MoO4比Table 2 Ni/K2MoO4 ratios of the K-Ni-Mo-based catalysts
2.6 H2-TPD分析
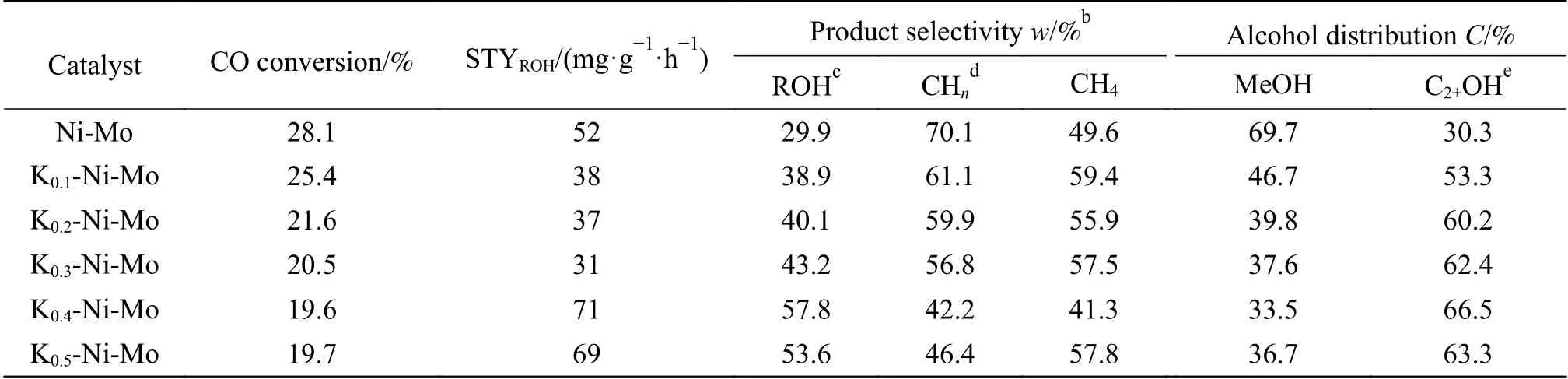
图8示出了具有不同K含量的K-Ni-Mo基催化剂的H2-TPD谱图。各K-Ni-Mo基催化剂在200–600 ℃均出现两种H2脱附峰。在250–450 ℃的低温峰I可归属于H物种在Ni表面的化学吸附,而在450–650 ℃的高温峰II可归属于H物种在Mo表面的强化学吸附[28]。如图所示,随着K含量的增加,催化剂的H2低温脱附峰I面积呈增大趋势,对应峰面积大小顺序依次为Ni-Mo < K0.1-Ni-Mo < K0.2-Ni-Mo < K0.3-Ni-Mo < K0.4-Ni-Mo 图8 不同K-Ni-Mo基催化剂的H2-TPD谱图Figure 8 H2-TPD profiles of different K-Ni-Mo-based catalysts 图9示出了具有不同K含量的K-Ni-Mo基催化剂的CO-TPD谱图。如图所示,除无K助剂的Ni-Mo催化剂外,各K-Ni-Mo催化剂在300–500 ℃均可观察到CO的脱附峰。在合成气制低碳醇反应过程中,催化剂表面吸附的非解离CO是参与成醇反应的关键物种[38]。由图9可知,在无K助剂的Ni-Mo基催化剂上未观察到非解离CO的脱附峰,而伴随K助剂的引入及其含量的提高,非解离CO脱附峰面积逐渐增大并且脱附温度向低温移动。上述研究结果表明,引入K助剂后K-Ni-Mo基催化剂上CO吸附量增高,对CO分子非解离活化的能力显著提升,进而有利于成醇反应的进行,这与不同K-Ni-Mo基催化剂的CO加氢成醇反应结果相一致,即各催化剂总醇选择性变化与K含量呈正相关趋势。 图10示出了具有不同K含量的K-Ni-Mo基催化剂的CO2-TPD谱图。Ni-Mo基催化剂上仅观察到在100–200 ℃的单一CO2脱附峰,该CO2脱附峰归属于催化剂上弱CO2化学吸附,表明Ni-Mo基催化剂上碱性位较弱。而在K-Mo基催化剂上观察到峰值位于450 ℃左右的CO2脱附峰,证明K-Mo基催化剂上具有强碱性位,对应于对CO2的强化学吸附[39]。由图可知,随着K含量的提高,不同K含量的K-Ni-Mo基催化剂上低温CO2脱附峰面积逐渐减小,而高温CO2脱附峰逐渐增大。已有研究结果表明,催化剂碱性增强有利于醇类产物的生成,但过强的碱性位会促进甲醇的生成,进而降低了C2+醇收率[40]。因此,本研究中可以通过K的引入来针对性调变催化剂碱性位,进而有效提高C2+醇选择性。 图10 不同K-Ni-Mo基催化剂的CO2-TPD谱图Figure 10 CO2-TPD profiles of the different K-Ni-Mo-based catalysts 对具有不同K含量的K-Ni-Mo基催化剂进行合成气制低碳醇反应性能的评价,结果如表3所示。传统Mo基催化剂通常在反应温度高于350 ℃、反应压力大于8 MPa时才能具有较高的CO加氢活性[41],而本实验中在240 ℃、5 MPa的相对温和条件下,CO转化率即可达到20.0%–25.4%。另外,也发现不同K含量的K-Ni-Mo基催化剂上CO转化率并无显著差异。尽管增加K引入量会导致催化剂上部分活性中心被覆盖[18],并且如表1所示的BET表征结果,观察到K含量提高同时也引起催化剂比表面积下降。但结合XRD及H2-TPD表征结果可知,引入K促进了K2MoO4相的生成,进而降低了NiMoO4含量,使还原后催化剂中加氢活性高的Ni物种含量提高。同时,根据表2中Ni/K2MoO4比的变化可以发现,各K-Ni-Mo基催化剂中Ni/K2MoO4比会随着K含量的增加表现为先增大后略微减小的变化趋势,也证明了K的引入有效促进了Ni物种的还原,这与以上XRD及H2-TPD分析结果相印证。因此,尽管K含量增大,但CO转化率并无明显降低。此外,如表2所示,研究也发现可以通过改变K引入量来实现调变Ni与K2MoO4之间的相对含量。其中,在K0.4-Ni-Mo催化剂上Ni/K2MoO4比最高,这显示存在最优的K引入量。对比表3中各催化剂反应评价结果,最佳的Ni/K2MoO4比出现在K0.4-Ni-Mo催化剂上,该K含量下的K-Ni-Mo基催化剂表现出最优的合成低碳醇反应性能。 表3 不同K-Ni-Mo基催化剂上低碳醇合成反应性能aTable 3 Catalytic performances of the K-Ni-Mo-based catalystsa 不同K-Ni-Mo基催化剂上总醇选择性随K含量的升高表现出先增大后降低的趋势。其中,K0.4-Ni-Mo基催化剂具有最优的总醇选择性(57.8%),是相同反应条件下Ni-Mo催化剂(29.9%)的近2倍。根据已有报道,CO非离解活化对于醇类产物的形成至关重要[42]。如图9所示CO-TPD表征结果可知,Ni-Mo催化剂对CO以解离活化为主,而非离解活化能力不足,从而更利于烃类产物生成,表现出较低的总醇选择性,这与反应评价结果相一致。基于CO-TPD分析结果,K-Ni-Mo基催化剂上更易发生的CO非解离吸附,并且非解离CO吸附量随K引入量的增加而提高。因此,引入K显著提升了K-Ni-Mo基催化剂上CO非解离吸附活化能力,而非解离CO吸附量的有效提升是KNi-Mo基催化剂上总醇选择性明显提高的关键因素。 此外,文献证实中/强碱性位在CO加氢生成低碳醇过程中也起到关键作用[43]。如图10所示,随着K含量的增加,不同K含量的K-Ni-Mo基催化剂上低温CO2脱附峰面积(弱碱性位数量)逐渐减少,而高温CO2脱附峰面积(强碱性位数量)逐渐增加。如前所述,尽管K引入量增大并未显著提高CO加氢活性,但K的引入增加了催化剂表面碱性,进而提升了催化剂上碱性羟基基团数量,从而降低烃类生成,这也与本研究中反应评价结果相一致[44]。研究显示,催化剂表面羟基与烷基金属–碳键的加成反应有利于醇产物的生成[19],因而显著提高了C2+醇选择性。但已有研究结果也显示出过强的碱性会促进甲醇的生成,进而降低C2+醇时空收率。因此,K-Ni-Mo基催化剂上存在最佳的K引入量。在K0.4-Ni-Mo基催化剂上总醇中C2+醇的选择性达到最高的66.5%,对应醇时空收率为71 mg/(g·h)。 在合成气制低碳醇研究中,CO插入机理已被广泛认可[38,45]。非硫化态K-Ni-Mo基催化剂上发生CO加氢成醇反应需要同时具有CO解离吸附和CO非解离活化的双活性位。在Ni活性中心上主要发生CO解离即碳氧键断裂和碳链增长反应,而K2MoO4相上发生CO非解离活化与插入成醇类反应。如图11所示,在反应过程中各烷基中间体CxHy可以通过CH2聚合发生碳链增长而形成,进一步加氢产生烷烃或通过脱氢生成烯烃;同时,各烷基中间体CxHy与非解离CO发生插入反应首先转化成酰基,继而发生加氢反应产生醇类产物[46],即Ni与K2MoO4物相间的双中心协同作用是非硫化态K-Ni-Mo基催化剂具有优异成醇性能的关键。 图11 K-Ni-Mo基催化剂上合成气制低碳醇反应网络图Figure 11 Schematic diagram of the reaction network over the K-Ni-Mo-based catalysts for higher alcohol synthesis from syngas 通过水热还原法制备出具有金属Ni和K2MoO4紧密接触的非硫化态K-Ni-Mo基催化剂。研究表明,K引入后生成了K2MoO4相,同时降低了NiMoO4相含量,促进了金属Ni的生成,进而明显提升了催化剂低温加氢活性和低碳醇选择性。另外,引入K增加了催化剂碱性,提高了催化剂表面碱性羟基基团数量,使得烃类产物选择性降低。其中,K0.4-Ni-Mo基催化剂具有最佳的催化性能。在5 MPa、240 ℃、空速5000 h-1的相对温和条件下,CO转化率达到19.6%,总醇选择性可达57.8%,C2+醇占总醇约66.5%,对应醇时空收率为71 mg/(g·h)。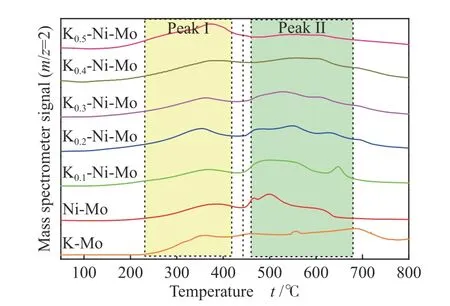
2.7 CO-TPD分析
2.8 CO2-TPD分析

2.9 不同K含量的K-Ni-Mo基催化剂上低碳醇合成反应性能

3 结 论
猜你喜欢
分子催化(2022年1期)2022-11-02
纺织标准与质量(2022年3期)2022-08-10
纺织标准与质量(2022年2期)2022-07-12
电气技术(2022年5期)2022-05-23
汽车工程师(2021年12期)2022-01-18
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
第一财经(2019年8期)2019-08-26
山东工业技术(2018年10期)2018-06-26
作文·初中版(2017年6期)2017-06-16
计算机辅助工程(2014年4期)2014-09-18
