
基于密度泛函理论的高覆盖氧吸附焦炭氧化机理研究
2022-09-09 02:23刘治港田向红李言钦
燃料化学学报 2022年8期
刘治港 ,田向红 ,李言钦,*
(1.郑州大学 机械与动力工程学院, 河南郑州 450001;2.郑州大学 化工学院, 河南郑州 450001)
碳的氧化燃烧不仅理论上是一个典型的复杂异相反应,而且在能源、化工、冶金、材料等方面有重要的工业应用意义。煤炭燃烧发电约占全球电力生产的40%,而微观上看,焦炭燃烧占煤粉整个燃烧过程的一半以上[1]。为了合理利用燃料,提高效率并减少对环境的影响,需充分理解反应气体在固体碳表面吸附、反应和脱附的微观机理,能够让人们更好的认识焦炭颗粒燃烧所发生的反应过程,因此,对其反应机理的研究非常重要,同时可为焦炭气化等反应的研究提供一定参考。焦炭的贮存也是人们所关心的重点,其氧化会对煤焦的工业特性产生影响;另一方面,贮存期间防止自燃同样十分重要[2],低温下焦炭表面部分区域往往有较高的氧吸附覆盖率,理解相应条件下的氧化反应机理很重要。
长期以来,人们对焦炭化学反应的解释主要关注其表面氧化物的形成和分解。李金虎[3]在对于焦炭表面活性位点产生及焦炭低温自燃抑制等方向做了相应工作。Zhu等[4]综述了焦炭表面氧化过程相关工作,其中,假设碳的气化是由表面环氧化复合物的形成促进的,仅仅基于化学热力学的研究,没有考虑过程的化学动力学性质。Montoya等[5,6]分别基于Armchair和Zigzag模型研究了CO2、H2O的焦炭气化相关机理,文献[7]则研究了CO在炭材料表面发生化学反应的动力学机理。Backreedy等[8]研究了一种在Zigzag焦炭构型边缘发生碳气化的机理,但没有提供有关动力学信息,其结论完全基于反应中键长的计算与分析。
Zhuang等[9]采用瞬态动力学和TPD方法研究了碳在气化过程中形成的表面氧络合物动力学行为,但其结果不涉及具体的化学动力学机理。Sendt等[10,11]研究了O2在Zigzag焦炭结构上吸附,产生羰基在反应表面迁移,进而生成CO的现象,而对于可能的CO2产生则没有讨论。文献[12]研究了CO2在Zigzag表面吸附的特性,但对于脱附反应过程并没有涉及。钟俊等[13]对Zigzag煤焦表面异相还原N2O反应进行了相应计算。Chen等[14]研究了H2O/Ar氛围下钠在炭表面的还原作用。Yang等[15]研究了木质素热解与炭化反应的机理。Frankcombe等[16]证实了氧原子在模型表面重排路径能量相对低,这也在文献[17]中得到了验证。Zhang等[18]利用相似模型基于DFT理论研究了C(N)-NO的反应。
另外,周赛等[19]研究了含氮Armchair焦炭模型在吸附CO2之后生成NO的反应机理。Orrego等[20]讨论了以萘氧基为模型体系的一些实验观察到的形成CO2的反应途径。其工作涉及氧原子从基面到边缘的转移过程,这与本研究不同。此外,Sánchez等[21]还研究了碳氧化反应中二氧化碳的演化过程,但他们的研究仅仅集中在CO/O2在焦炭再吸附之后的CO2生成现象。Radovic[22]同样研究了碳氧化过程中分别为直接方式和间接方式的二氧化碳脱附,但他们所做工作中没有考虑模型表面C-O团簇的迁移以及由此生成CO2的可能路径。
总之,由于碳氧化/气化机理的复杂性,同时微观实验条件的局限,目前,关于相关的研究并不全面,尚没有得到完全证实的反应机理。因此,有必要从不同角度研究反应的相关机理,以期对现有研究形成补充,更好、更充分地理解相关的内在机理。鉴于当前能源高效清洁利用的要求愈发迫切,低温条件下(900 K以下)[23]或焦炭氧化/燃烧可获得相对高的能量效率,其相应于高覆盖率氧吸附条件,后者也是一种较高压条件下的碳氧化场景。本研究以该条件下的焦炭氧化机理为对象,基于密度泛函理论研究了相应炭表面氧化生成CO/CO2的相关机理。
1 模拟计算
1.1 模型选择
Zhuang等[9]选用由六个苯环组成的Zigzag模型和Armchair模型来模拟焦炭氧化反应,得到了与实验相吻合的结果。同时,田向红[24]通过理论计算也得出Zigzag模型边缘位点相比Armchair模型具有更高的化学活性,本研究选用七个苯环的Zigzag模型,来模拟焦炭氧化反应的机理。具体模型结构如图1所示。对于该苯环簇结构,余留四个未饱和的碳原子模拟活性位,即图1中C2、C4、C6和C8,其他位置碳原子利用H原子封闭。图2所示为Zigzag焦炭表面满覆盖氧吸附模型。

图1 Zigzag与Armchair焦炭表面模型示意图Figure 1 Zigzag (left) and Armchair (right) models of coke molecule
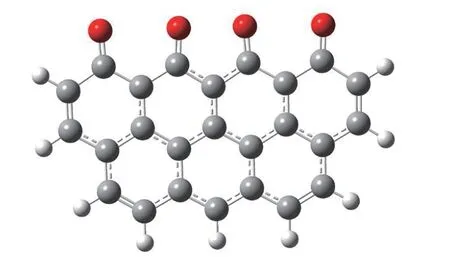
图2 高覆盖度Zigzag表面氧吸附模型示意图Figure 2 Oxygen adsorption model with high coverage along the Zigzag coke edge
1.2 计算方法
模型中采用DFT/B3LYP方法和6-31G(d)基组进行结构优化和频率分析。根据不同自旋多重度下电子能量比较之后,选取能量最低的双重度作为模型自旋多重度,基于此,反应物、中间体、过渡态和产物都得到了充分优化。每个过渡态结构对应一虚频,其原子振动方向与反应趋势一致,另进行了IRC路径分析验证[25],以确保反应发生的正确性。为保证计算结果准确性,选取高精度基组6-311++G(d, p)进行单点能的计算[26],其中,需要考虑能量矫正,这里根据所使用的计算基组采用相应经验值零点能矫正因子0.98,如文献[17]基于该值研究了汞选择性催化非均相反应相关机理。
1.3 模型验证
首先,对所建立焦炭氧化的DFT模型以及计算基组的可靠性进行验证。基于可比性,计算了Zigzag活性表面处吸附O2分子以及表面CO脱附过程,将计算结果中各步吉布斯自由能能垒与文献[9]和[10]结果进行对比,如图3所示,其中,TS1与TS2对应为O2吸附过程过渡态能垒值,TS3-TS6以及PTS1-PTS2分别为后续第一、二个羰基脱附过程。计算方法与模型得到基本的可行与可靠性验证。基于此,本研究进一步建模探究了Zigzag焦炭表面CO/CO2生成并脱附的反应路径。
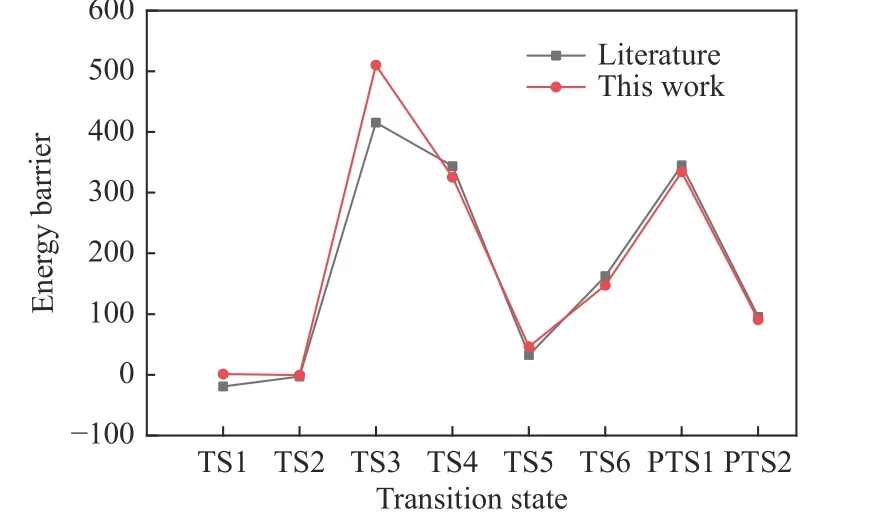
图3 焦炭氧化基本反应过程计算与文献[9,10]对比Figure 3 Comparison between the calculation results and the literature[9,10]with respect to the basic reaction process of coke oxidation
2 结果与讨论
2.1 边缘位点碳氧基团脱附
基于图2吸附模型,考虑两种典型情况的脱附,第一种是模型活性区边缘处(图1中C2/C8位点)CO/CO2的脱附,相应在该处失掉一个C形成
五元环结构;第二种则为活性区内部C4/C6位点处的CO/CO2脱离,在该处最后形成五元环。
首先讨论第一种,计算结果相应构型变化如图4所示,这里以符号X表示全覆盖氧吸附态,为反应过程初始态,Z表示由C2或C8形成的羰基的脱附过程(基于模型的对称性),其他过程的中间体这里命名为为An(n= 1、2...),ts表示过渡态。初始构型经过渡态Ats,会形成稳定的中间产物A1。这一过程需克服196.65 kJ/mol的能垒,吸收159.41 kJ/mol的热量。然后C9-C7键形成,键长1.573 Å。C8-C9键和C8-C7键的长度分别为1.476 和1.550 Å。之后C8-C7键断裂,形成过渡态A1ts。由图4可以看出,C8-C7键伸长为1.914 Å,随着C8-C7键断裂A2形成。这一过程需要克服14.40 kJ/mol的能垒,吸收2.04 kJ/mol的热量。由构型A2,CO可以通过过渡态Zts直接脱附,Zts处的能垒为8.28 kJ/mol,该过程释放21.45 kJ/mol的热量。同时,通过过渡态A2ts,C8将与另一个O(C6位置)连接形成一个五元环,即为A3构型,这一过程需要跨越2.34 kJ/mol的能垒,结束后释放27.06 kJ/mol的热量。随后原来的C8-C9键变为1.565 Å,很容易断裂,通过A3ts,形成A4中间体。A3→A4反应需要克服112.38 kJ/mol的能垒并释放17.58 kJ/mol的热量。通过过渡态A4ts,形成更稳定的中间体A5,需要克服52.97 kJ/mol的能垒,释放出165.06 kJ/mol的热量。A5的形成始于C-O的团簇转移到活性表面中部,然后与另外两个O原子(C4和C6位置)形成C-O键,形成一六元闭环结构,其能量比初始构型X更低。
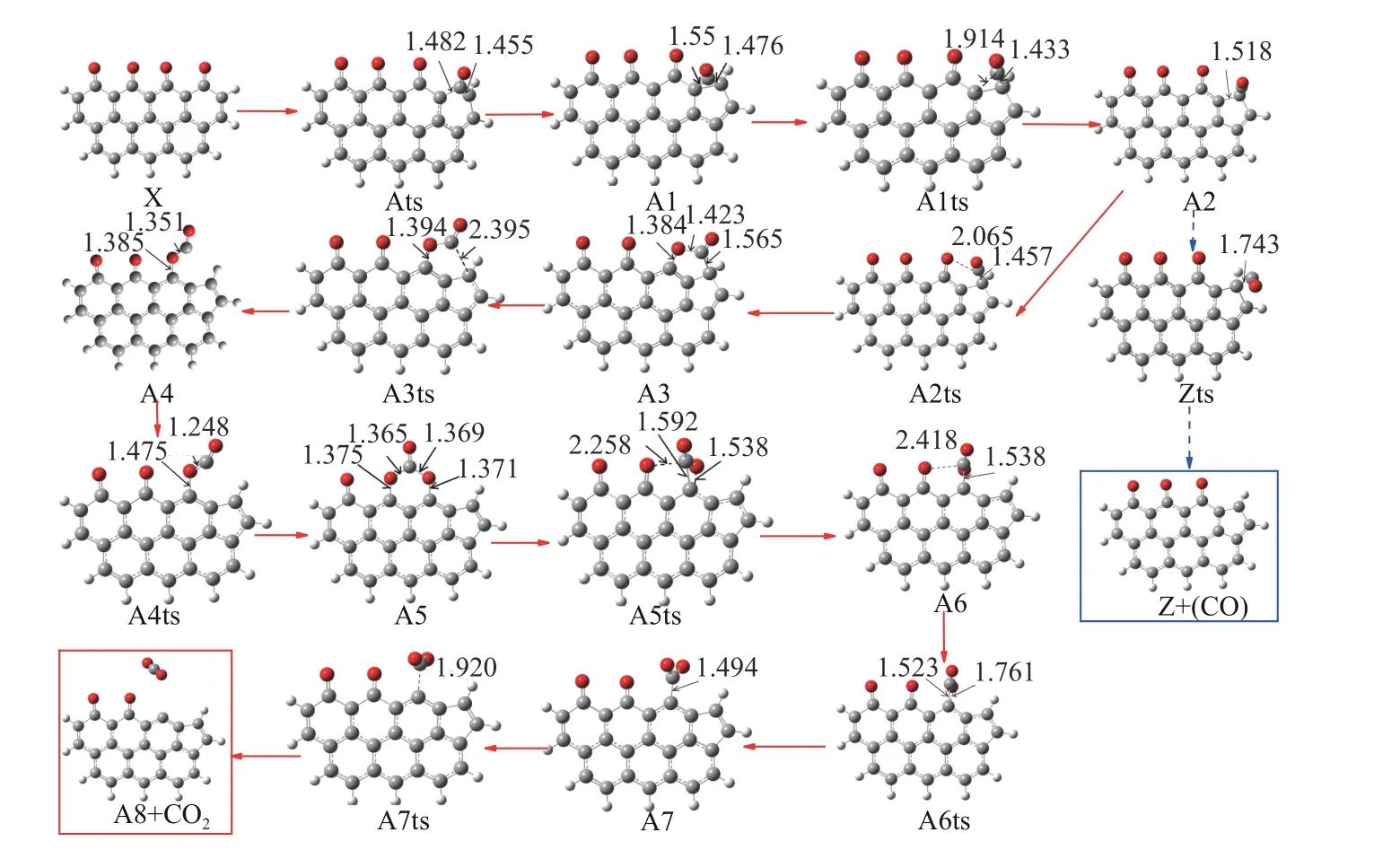
图4 模型边缘活性位CO/CO2的生成与脱附过程对应构型变化(所框为路径最终产物)Figure 4 Corresponding configuration changes of the desorption of CO/CO2 at the edge of the coke molecule
C8和C6之间的振动形成过渡态A5ts,其键长是1.592 Å。在克服323.74 kJ/mol的能垒,吸收309.16 kJ/mol的热量后,另一中间体A6形成,C8-C6键长变为1.538 Å。A5ts构型上的二面角结构O-C8-C6-C发生扭转成为构型A6。其进一步扭转形成A7。此时连接到C8的两个O原子,几乎落在同一平面上,相应的能垒为6.65 kJ/mol,释放热量为57.24 kJ/mol。最后,C8和C6之间的振动使它们之间的键断开,从而使形成CO2并脱附。相应过渡态为A7ts,其中,C8-C6的键长为1.92 Å,该步能垒为34.73 kJ/mol,整个过程释放的热量为35.15 kJ/mol。图4反应过程相应势能面见图5,详细动力学参数见表1。
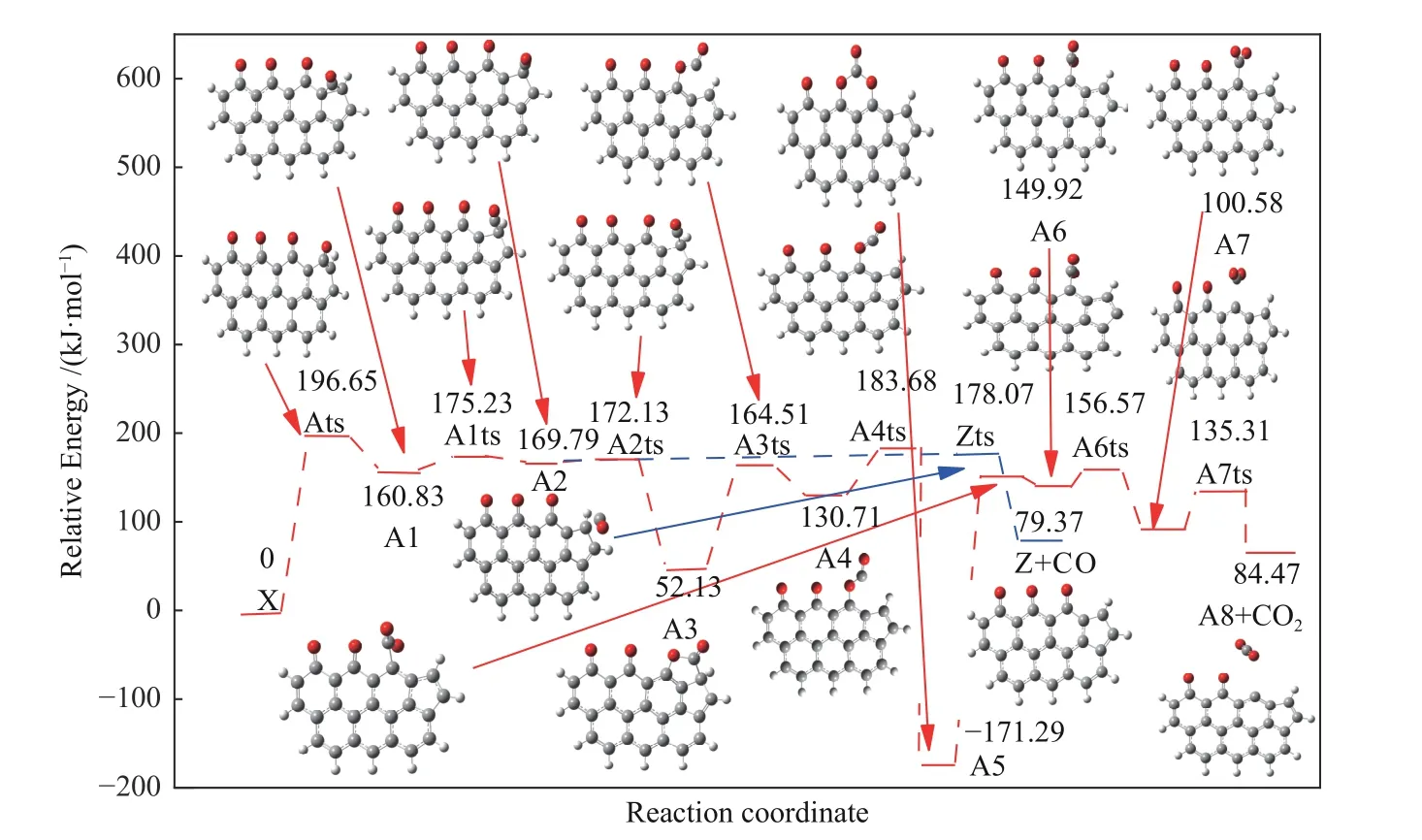
图5 边缘活性位CO/CO2的脱附过程相对能量变化Figure 5 Diagram of relative energy with CO/CO2 desorption at the edge active sites
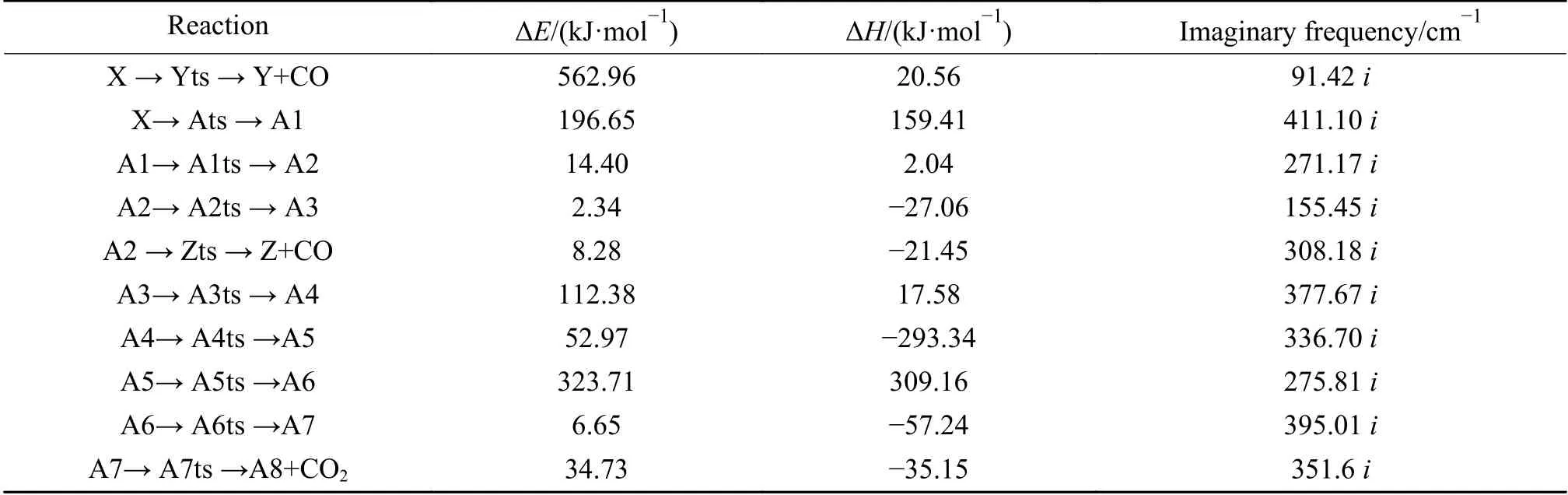
表1 边缘活性位CO/CO2的生成与脱附过程的能垒、焓变及过渡态虚频Table 1 Energy barrier, enthalpy change and imaginary frequency of each step in the CO/CO2 formation and desorption process
从反应路径所经势能面看,该部分反应路径初始步能垒较高,对应于边缘位置处六元苯环缩聚为五元环过程。另外羰基可直接脱附或与临近氧原子相连形成碳氧五元环结构,两者均对应较低的能垒。后者重排后形成较稳定的A3结构,值得注意的是,重排反应步能垒很低,且其对应的过渡态能量均比初始步过渡态能量低,意味着后续正向反应比逆向反应更容易发生。随后A3结构中五元碳氧环易被破坏并继续重排,羰基迁移,形成更为稳定的碳氧六元环结构A5。随后碳氧六元环在克服很高的能垒后发生破坏,但其过渡态对应能量仍比初始步较低,因此,即使该步存在较高能垒,但并不显著影响反应继续进行。后续过程分子经过两个重排过程,能垒最高不超过50 kJ/mol,最终CO2以与焦炭分子异面的形式完成脱附,综合来看初始步过渡态能量最高,其对反应影响最为重要。
2.2 中间位点碳氧基团脱附
此外,该全覆盖吸附模型可能有另一种反应路径,即前述第二种反应可能。鉴于这里的结构对称性,以左半部分为例,C3-C4键可能断裂,反应通过数个过渡态和中间体以不同的方式进行,最终脱附CO或CO2。图6、7以及图8、9分别显示了不同路径的整个过程构型和反应势能面的相应变化。在该过程中Y表示CO (在C6位置处)的直接脱附,U表示CO间接脱附。其余过程各驻点相应构型用Bn(n= 1、2....)表示,ts用以表示过渡态。
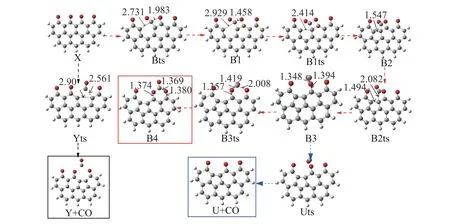
图6 初始构型X分别到B4及U+CO(图中所框为过程末端构型)过程的构型变化Figure 6 Configuration diagram of the initial configuration X to B4 and CO, respectively, with the frames denoting the final configurations (the frame is the final configuration)
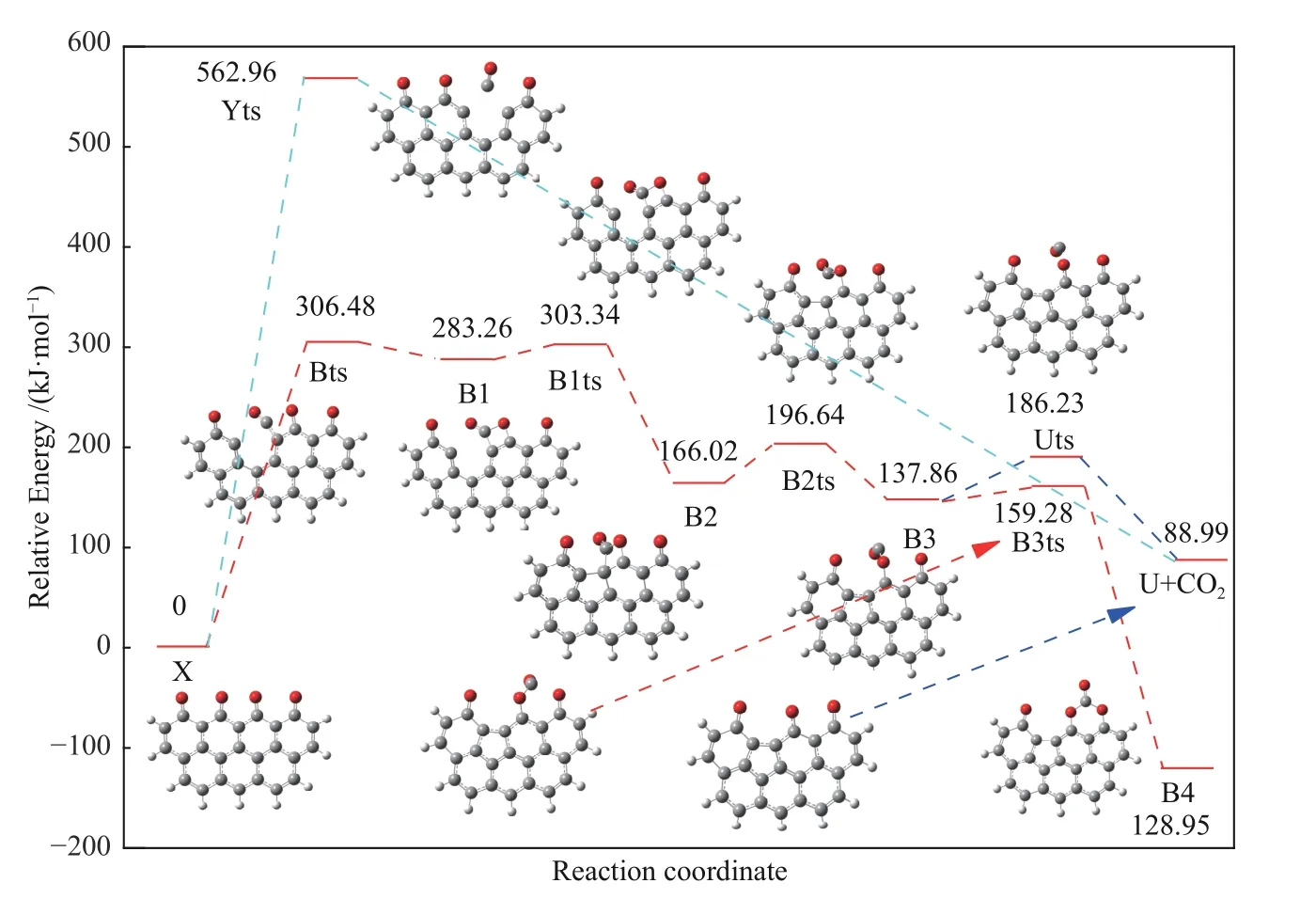
图8 初始吸附构型重排至B4过程相对能量变化Figure 8 Diagram of relative energy change from initial configuration rearrangement to B4
由图6和图8可知,CO可以直接脱附,对应的过渡态为Yts,其需要克服相当大的能垒562.96 kJ/mol,整个过程将吸收20.56 kJ/mol的热量。此外,初始构型中C3-C4键断裂,形成过渡态Bts,能垒为306.48 kJ/mol,相应的C4与C6上的O结合成键,吸收热量为271.29 kJ/mol。然后C3和C5通过B1ts连接,能垒为20.08 kJ/mol,该过程会释放103.93 kJ/mol热量并生成中间体B2。由于C3-C5键的形成,C4-C5键容易断裂,对应于过渡态B2ts(能垒为30.62 kJ/mol),B2到B3的过程放出38.95 kJ/mol的热量。此时,C4和O原子之间的振动使CO脱附容易发生,对应过渡态Uts(能垒48.37 kJ/mol),产物为CO和U(释放71.72 kJ/mol热量)。通过过渡态B3ts(能垒为21.42 kJ/mol),原子C4再与C8上的O原子连接,形成更稳定的中间体B4(释放热量为247.52 kJ/mol)。此时,二氧化碳可以三种相似的方式脱附,分别从C2、C6或C8三个不同位点脱附,如图7。
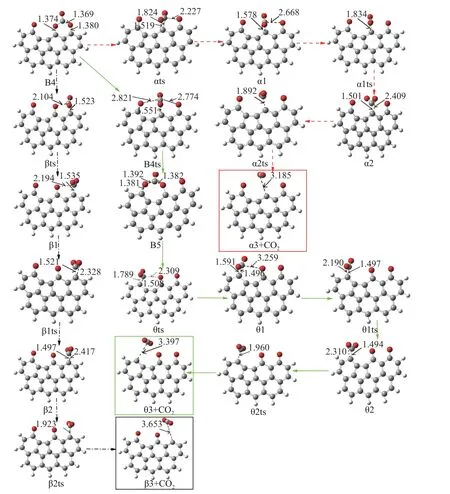
图7 由B4到CO2脱附的各路径构型变化(框内为相应路径产物)Figure 7 Configuration changes of each path from B4 to CO2 desorption (the products of each path in the color boxes)
构型B4通过过渡态 B4ts 转化为B5,同时C4与C2位置的O原子相连。相应能垒为259.37 kJ/mol,需吸收热86.15 kJ/mol。如图6与图7所示,B5与B4两个构型相似,在最外层有四个碳原子和两个氧原子形成一个闭环结构(六元环)。在这些结构的基础上,C4-C6、C4-C8(中间体B4)和C3-C2(中间体B5)的振动使中间体α1、β1和θ1的形成成为可能,相应过渡态分别为αts、βts和θts,分别具有281.83、295.26和228.19 kJ/mol的能垒。该部分的动力学参数详见表2。随后基团O-C4-C角度发生扭转,最终CO2-C结构中的两个氧原子处于几乎同一高度,该部分反应同前述第一部分边缘位点CO2脱附相似,需经历较小的能垒扭转形成中间体α2、β2和θ2,最终通过CO2-C结构中C-C键断裂的形式完成CO2脱附过程,该部分反应相对自由能变化见图9,相关参数见表2。
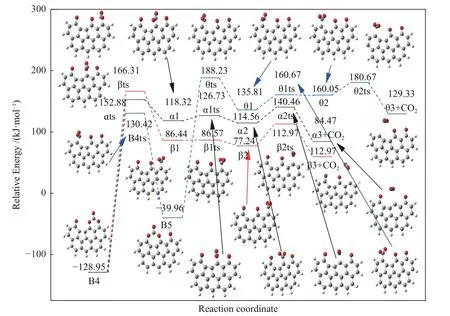
图9 碳氧六元环结构B4重排脱附CO2过程的相对能量变化Figure 9 Relative energy diagram of six-membered ring rearrangement to CO2 desorption
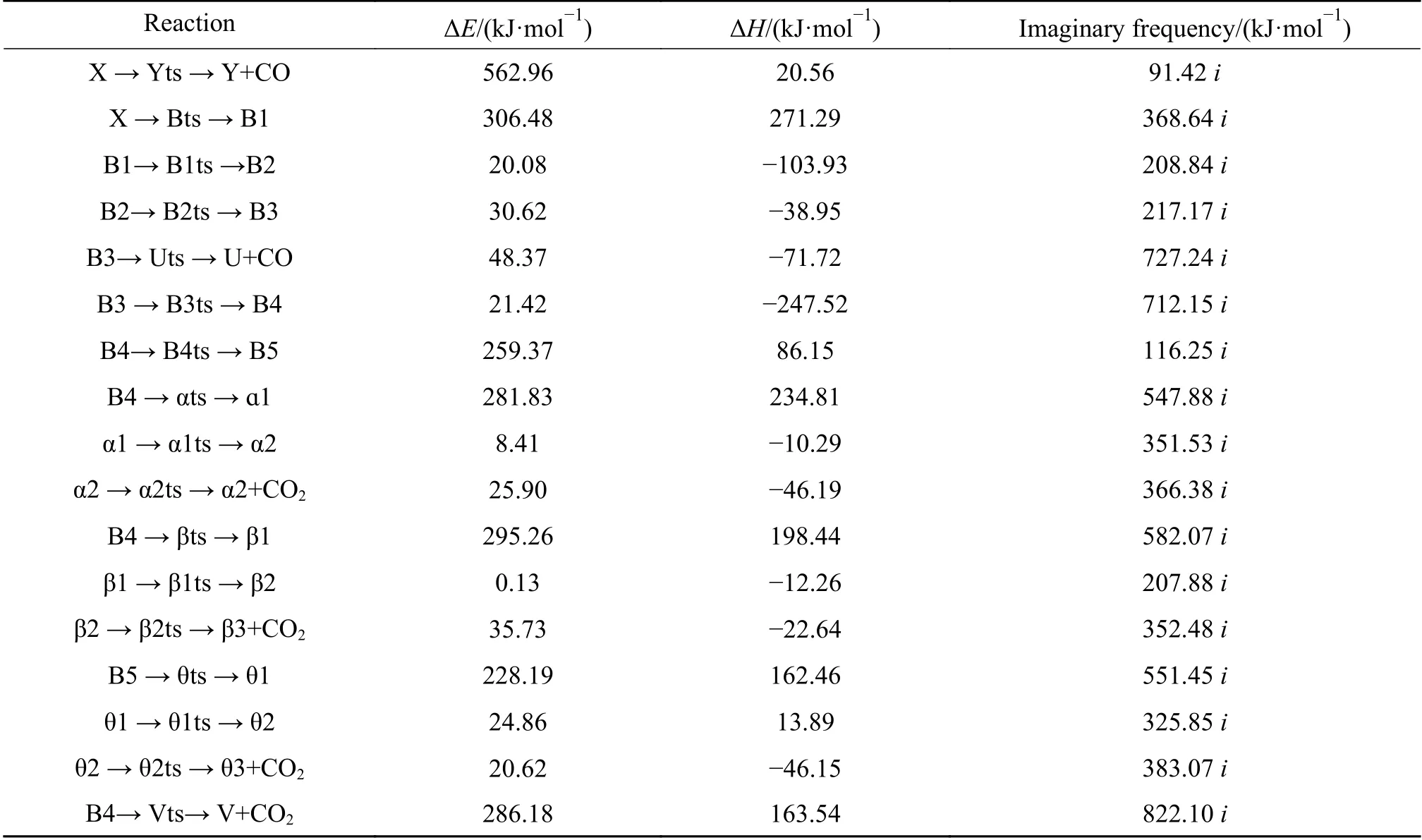
表2 中间位点重排及脱附过程中各基元步的能垒、焓变以及虚频Table 2 Energy Barrier, enthalpy change and imaginary frequency of each step in the processes of rearrangement and desorption
另外,除上述以最终C-C键断裂为特征的反应路径,CO2脱附过程也可以从中间体B4以一种直接断掉两个C-O键的一步反应方式实现,具体形式见图10,相对自由能变化见图11。可以看到从具有C-O六元环结构的中间体B4,O-C4-O基团可以CO2的形式直接脱附,表现为C4-O以及C8-O键的同时断裂,相应的键长分别伸长至2.078和1.966 Å。该过程对应的过渡态记为Vts,其后的产物以V+CO2表示。该步反应的能垒为281.68 kJ/mol,相应焓变为163.54 kJ/mol,该过程的相对自由能变化见图11。该机理相关但不同于文献[21]逐个断掉C-O键而脱附CO2的反应方式。

图10 上述六元环结构直接脱附CO2过程构型变化Figure 10 Configuration diagram of direct desorption of CO2 from a six-membered ring structure

图11 图10反应过程相对能量变化Figure 11 Diagram on relative energy of direct desorption of CO2 related to Figure 10
从整体反应势能图看,该部分反应初始步对应能垒较高,这与前述2.1部分类似,从图8中可以看到,羰基直接脱附需要跨越高能垒。在重排至形成碳氧六元环结构B4的过程中,后续基元步的能垒与初始步相比很小,最高的不足40 kJ/mol,随后的继续重排至脱附过程中,单步对应能垒较高,为260-300 kJ/mol,但其在相对势能面中过渡态能量最高只有166.31 kJ/mol,明显小于初始步306.48 kJ/mol。值得一提的是,θts对应的基元步能垒与前述αts、βts相比较低,这是因为B5与B4结构的不同,B5对应形成碳氧七元环结构,而六元环中分子间距与之相比更为紧凑,因此,七元环对应结构稳定性较弱,在翻越较小的能垒后完成重排。上述过程后续重排至脱附过程对应能垒很小,反应很容易正向进行。而Vts对应的CO2脱附过程相对简单,反应仅需经由单步克服286.18 kJ/mol的能垒即完成脱附,这与前述几种重排至CO2基团并与模型分子异面结构完成脱附对应最高能垒相近,即其反应难易程度相近。
最后,将Sanchez等[21]工作中CO2脱附方式与本文进行了对比,前者在模型表面吸附O2分子后,再吸附CO至边缘处氧原子之间得到闭合六元环结构,相应于本研究B4构型,随后模型表面的O-C键断裂而形成CO2-O-C的结构,最后由于CO2-O结构中C-O键的断裂,使得CO2脱附完成。其相应于本研究典型的CO2生成并脱附的反应路径B4 →...→ α2 + CO2,但不同的是,本研究中的CO2脱附通过断掉C-C键实现,两种反应路径均由多步基元反应组成。
2.3 反应动力学
基于前述计算结果,对所得各反应路径进行反应动力学分析。根据过渡态理论,在前述模型基础上,采用速控步方法计算了不同温度下CO/CO2脱附过程相应化学反应速率常数,见图12。通过曲线拟合阿伦尼乌斯方程,分别得到相应反应路径决速步指前因子和活化能。由式(1)计算并基于图12的曲线拟合,可得决速步活化能(具体见2.4节),须注意的是其通常与相应能垒不同[27]。
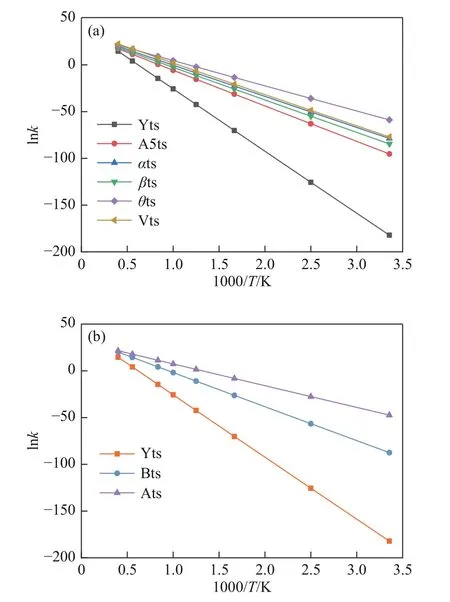
图12 各脱附反应路径速控步反应速率常数随温度的变化Figure 12 Diagram of rate constant of rate-controlled step vs temperature in each desorption pathway
由DFT计算结果确定反应速率常数[28]的方法如下:

式中,λ是量子轨道效应的修正因子,经验表达式为:

Ea是反应活化能,R是气体常数。kB是玻尔兹曼常数,h是普朗克常数,T为热力学温度。Q≠为过渡态的配分函数,QR为反应物的配分函数。作为完全配分函数,可由三个分量的乘积来表示:

式中,包括平动配分函数Qt、转动配分函数Qr和振动配分函数Qv。
从图12中可以看到,同等条件下CO直接脱附反应的反应速率常数明显低于其余反应过程,这说明CO直接脱附过程较难发生。由图12(a)可知,CO2的几种不同的脱附方式则有着相近的活化能,反应难易程度并无明显区别;值得注意的是,其中,θts所在的C-C键断裂的CO2脱附相对于C-O键断裂的Vts路径更易发生。图12(b)比较了几种CO脱附的不同反应路径的相对难易,在同一条件下CO直接脱附过程对应的Ats有着最高的反应速率常数,这意味着某种间接CO脱附过程相对CO2易发生。与Bts所决定路径相比,两者分别发生于边缘和中间位点,说明同等条件下焦炭活性表面边缘处CO的脱附反应相对易发生。
2.4 结果验证
上述1.3部分对本研究所基于DFT对模型的计算结果进行了基本的验证。基于此所建立的不同于文献的CO/CO2脱附反应路径进一步与文献实验所得动力学数据进行了比较,结果见表3。其中文献所使用实验方法分别为热重技术[23,29]和TPD方法[30]。可以看出,本研究模型计算所得高覆盖氧吸附条件下CO和CO2脱附反应活化能与不同文献结果中实验值吻合良好,而通过本研究模型研究,本研究得到了实验方法很难获得的相应内在反应机理。
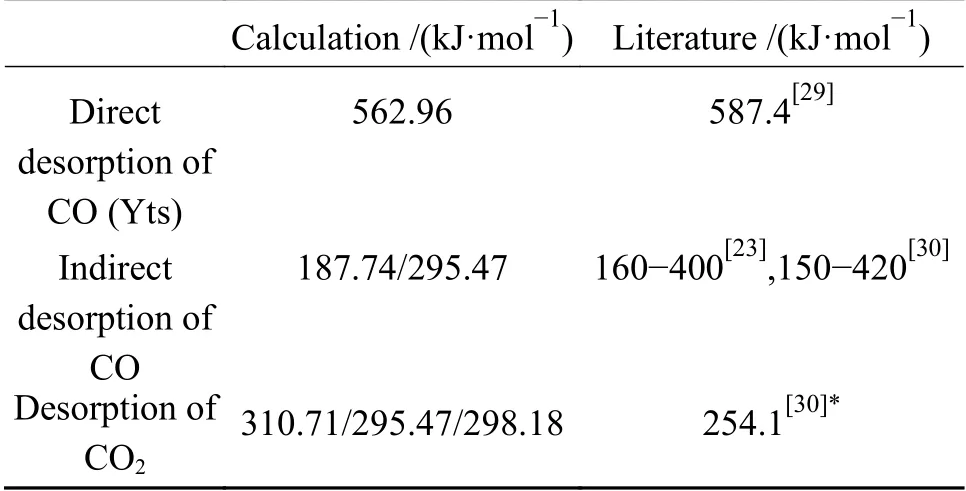
表3 脱附过程活化能与文献[23,29,30]对比Table 3 Calculated activation energies of desorptions compared with literature[23,29,30]
3 结 论
基于密度泛函理论,本实验研究了相应于较低温度或较高压力条件下生成的O2高覆盖率吸附焦炭表面脱附生成CO/CO2的机理,得到了CO直接与间接脱附不同过程。建立和获得了在相似焦炭构型条件下与文献不同的反应路径。尤其是CO2的脱附机理,文献相应反应路径为C-O键断裂而脱附产生CO2,而本实验所构建相应反应步骤则一方面得到了通过断裂C-C键完成CO2脱附的路径,另一方面获得了与文献不同两个C-O键同时断裂生成CO2的机理。本实验所得各典型脱附反应机理均得到相关文献实验及理论结果的合理验证,证明了本实验所提出反应路径的可行性。
猜你喜欢
矿山安全信息(2022年8期)2022-11-25
军民两用技术与产品(2022年1期)2022-06-01
西北工业大学学报(2022年1期)2022-04-22
矿山安全信息(2021年3期)2021-11-30
商品与质量(2021年7期)2021-11-23
昆钢科技(2021年3期)2021-08-23
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
当代陕西(2020年23期)2021-01-07
科技传播(2019年22期)2020-01-14
智富时代(2018年8期)2018-09-28
