
RP-HPLC测定复方双嘧达莫乳膏中3种主要成分含量
2022-09-08 10:24:12董宁霞李新新满玉清高俊玲
滨州医学院学报 2022年4期
董宁霞 李新新 满玉清 高俊玲
滨州医学院附属医院药学部 山东 滨州 256603
带状疱疹是由长期潜伏在脊髓后根神经节或颅神经节内的水痘-带状疱疹病毒(varicella-zoster virus,VZV)经再激活引起的感染性皮肤病,临床表现为单侧水泡疹并伴有神经性疼痛,常发病于年龄较大、免疫抑制或缺陷的人群,严重影响患者生活质量[1-2]。
复方双嘧达莫乳膏是一种复方外用乳膏制剂,可用于治疗带状疱疹,疗效显著且不良反应较小,其主要成分为双嘧达莫、消旋山莨菪碱和盐酸达克罗宁。该软膏通过对三种主要成分的联合应用,发挥软膏的抗疱疹病毒、镇痛消炎等药理作用。为保证复方双嘧达莫软膏质量的稳定可控,本研究建立了双嘧达莫、消旋山莨菪碱和盐酸达克罗宁的含量测定方法,为该软膏的质量控制提供了科学依据,同时也为含双嘧达莫、消旋山莨菪碱或盐酸达克罗宁等成分的药品含量测定提供参考。
1 仪器与材料
岛津LC-2010C HT(紫外检测器);InertSustain C18色谱柱(4.6 mm×250 mm,6 μm);FA2204B电子天平(上海精密科学仪器有限公司);AUW220D 型分析天平(日本岛津);KQ-100型超声波清洗器(昆山市超声仪器有限公司)。
复方双嘧达莫乳膏(自制,含双嘧达莫5%、盐酸达克罗宁0.5%、消旋山莨菪碱1%);双嘧达莫对照品、盐酸达克罗宁对照品、消旋山莨菪碱对照品(中国食品药品检定研究院,批号分别为100244-201003、100423-201102、100249-201303);甲醇为色谱纯,磷酸、磷酸二氢钾等试剂均为分析纯。
2 方法与结果
2.1 色谱条件 复方双嘧达莫乳膏含量测定的色谱条件见表1。
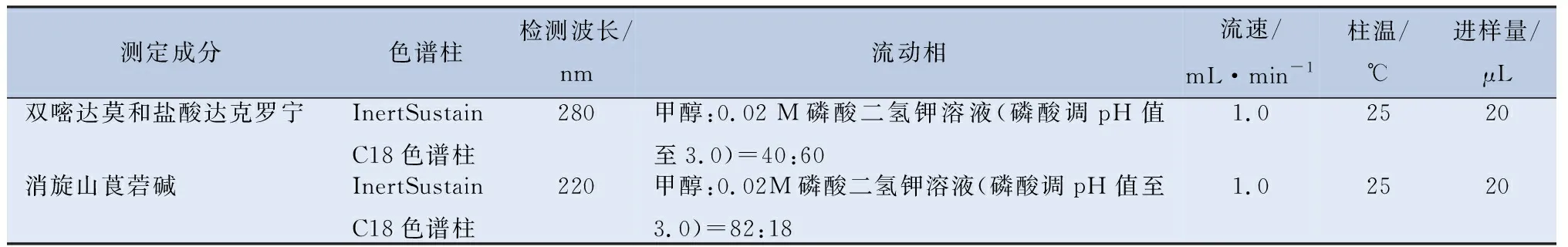
表1 复方双嘧达莫乳膏含量测定的色谱条件
2.2 溶液的制备
2.2.1 对照品储备液的制备 精密称取双嘧达莫对照品74.22 mg和盐酸达克罗宁对照品7.26 mg,置于同一50 mL量瓶中,用甲醇溶解并稀释至刻度,摇匀,即得双嘧达莫和盐酸达克罗宁混合对照品储备液。精密称取消旋山莨菪碱12.50 mg,放置于50 mL量瓶中,用甲醇溶解并稀释至刻度,摇匀,即得消旋山莨菪碱对照品储备液。
2.2.2 供试品溶液的制备 称取本品约0.4 g,加甲醇25 mL,超声处理15 min(功率为200 W,频率为44 kHz),冰浴冷却1 h,取出,迅速滤过,加甲醇定容到25 mL容量瓶中,作为供试品溶液,备用[3-4]。
2.2.3 空白样品溶液 按照复方双嘧达莫乳膏的处方工艺制备不含双嘧达莫、盐酸达克罗宁、消旋山莨菪碱原料药的空白样品,称取约0.4 g,按“2.2.2”项下方法操作,制备空白样品溶液,备用。
2.3 系统适用性试验 取“2.2.1”项下的对照品贮备液,稀释双嘧达莫浓度至742.2 μg/mL、盐酸达克罗宁浓度至72.6 μg/mL、消旋山莨菪碱浓度至120.5 μg/mL,对色谱系统进行适用性考察[5]。实验结果表明,理论板数按双嘧达莫、盐酸达克罗宁、左旋山莨菪碱、右旋山莨菪碱计,分别为10 904、12 286、8 104、8 231;双嘧达莫、盐酸达克罗宁、左旋山莨菪碱、右旋山莨菪碱的拖尾因子分别是1.09、1.10、1.06、1.06,色谱峰面积的相对标准偏差分别为1.3%、1.6%、1.7%、1.6%,分离度均大于1.5,色谱系统适用性良好。
2.4 方法学验证
2.4.1 准确度 精密称取空白基质约0.4 g,分别加入适量双嘧达莫、盐酸达克罗宁、消旋山莨菪碱对照品储备液,搅拌均匀,制成含双嘧达莫、盐酸达克罗宁、消旋山莨菪碱均为处方量100%的样品,平行配制6份。按“2.2.2”项下的方法制备供试品溶液,用0.45 μm微孔滤膜滤过,按“2.1”项下色谱条件进样,分别测定峰面积并计算回收率(表2~4)。
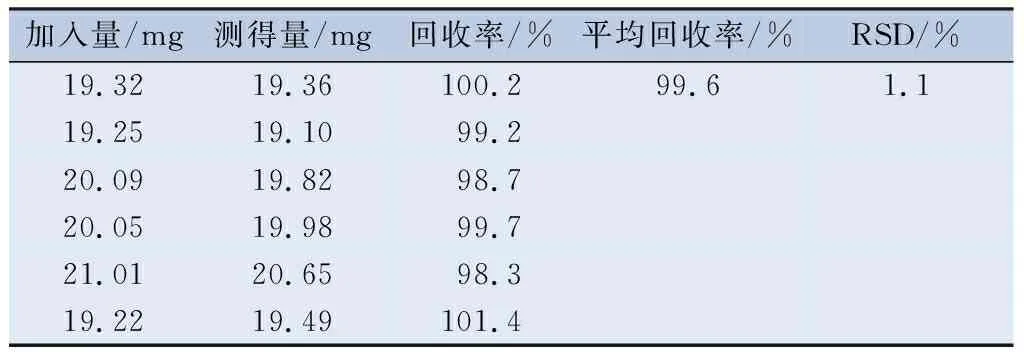
表2 双嘧达莫含量测定的回收率(n=6)

表3 盐酸达克罗宁含量测定的回收率(n=6)
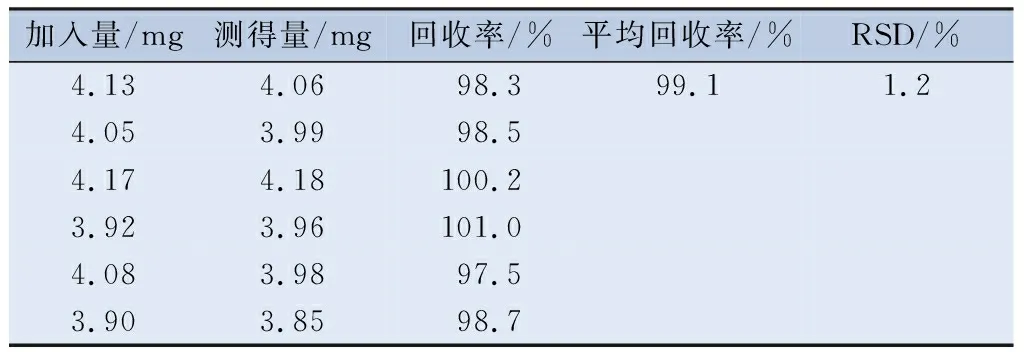
表4 消旋山莨菪碱含量测定的回收率(n=6)
2.4.2 精密度 同一份双嘧达莫乳膏,平行取样6次,每次0.4 g,精密称定。按“2.2.2”项下方法配制供试品溶液,用0.45 μm微孔滤膜滤过。按照“2.1”项下的色谱条件进样测定。结果表明,双嘧达莫、盐酸达克罗宁、消旋山莨菪碱含量测定的RSD(n=6)分别为1.8%、1.5%、1.8%,该测定方法的精密度良好。
2.4.3 线性与范围 精密吸取“2.2.1”项下的双嘧达莫和盐酸达克罗宁的混合对照品贮备液适量,添加流动相稀释,制备一系列线性关系工作液,使双嘧达莫和盐酸达克罗宁的浓度分别为247.4、494.8、989.6、1 237.0、1 484.4和24.2、48.4、96.8、121.0、145.2 μg/mL。按“2.1”项下色谱条件进样,分别测定峰面积。以峰面积y对进样浓度x进行回归处理,回归方程、线性、范围见表5。精密吸取“2.2.1”项下的消旋山莨菪碱对照品贮备液适量,添加流动相稀释,制备一系列线性关系工作液,使消旋山莨菪碱的浓度为50.0、62.5、125.0、187.5、250.0 μg/mL。按“2.1”项下色谱条件进样,记录色谱图,峰面积按消旋山莨菪碱顺、反式异构体峰面积的和计算。以峰面积y对进样浓度x进行回归处理,回归方程、线性、范围见表5。
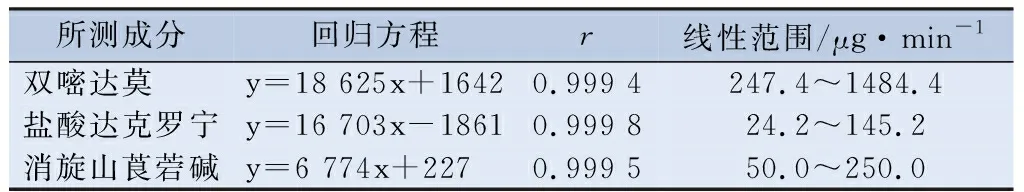
表5 复方双嘧达莫乳膏含量测定的线性回归方程
2.4.4 专属性 分别取“2.2”项下的空白样品溶液、供试品溶液、对照品储备液各适量,按照“2.1”项下的色谱条件进样,记录色谱图(图1、2)。空白基质及杂质不干扰双嘧达莫、盐酸达克罗宁、消旋山莨菪碱的含量测定。
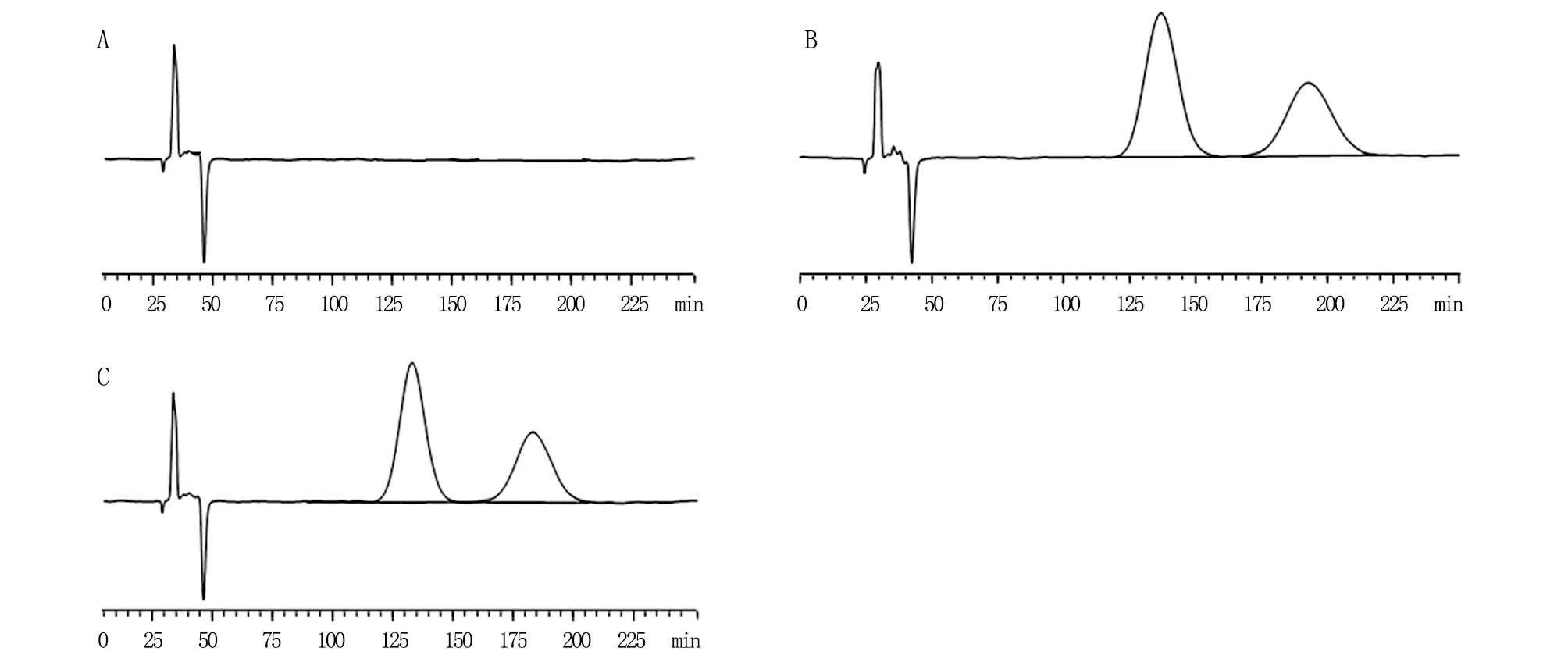
A.空白样品溶液;B.对照品溶液;C.供试品溶液。

A.空白样品溶液;B.对照品溶液;C.供试品溶液。1.盐酸达克罗宁;2.双嘧达莫。
2.4.5 耐用性 本研究分别考察流动相比例变化±5%、柱温变化±5℃、被测溶液放置0~72 h、流速相对值变化±20%时,仪器色谱行为的变化。试验结果显示:主峰与杂质峰可达到基线分离,主峰的拖尾因子小于1.5,各条件下的峰面积测量值(n=5)的相对标准差均小于2.0%。这表明,该色谱系统具有良好的耐用性。
2.5 含量测定 精密称取3个批次的复方双嘧达莫乳膏,每份约0.4 g,分别按“2.2”项下制备供试品溶液,用0.45 μm微孔滤膜滤过。按照“2.1”项下的色谱条件进样测定,用外标法计算含量(表6)。
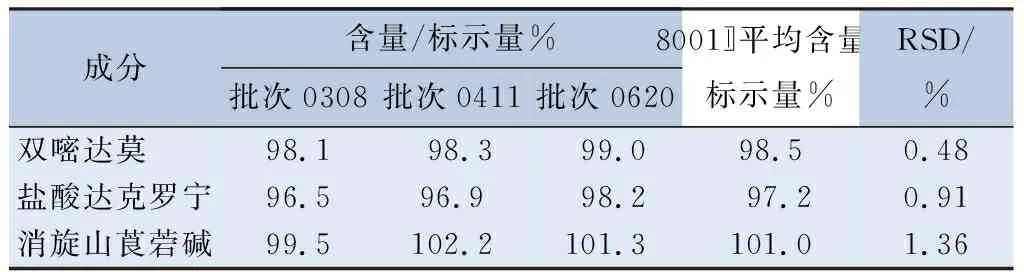
表6 复方双嘧达莫乳膏含量(n=3)
3 讨论
本研究的提取条件考察实验显示,选择超声提取时间为15 min时,可保证乳膏样品提取完全。但由于样品经超声提取时已近乳化状态,较难滤过,故将提取液冰浴1 h,可使提取液分层明显,易于过滤,以便进行下一步操作。
本研究用紫外分光光度计扫描各对照品溶液的全波长吸收光谱,发现双嘧达莫、盐酸达克罗宁在275~285 nm之间有最大吸收,而消旋山莨菪碱的最大吸收波长在220 nm左右。对275~285 nm波长范围进行重点考察,结果表明,在280 nm波长下,双嘧达莫、盐酸达克罗宁的峰高相对较高,且噪声小,故选择280 nm作为双嘧达莫、盐酸达克罗宁的检测波长,220 nm为消旋山莨菪碱的检测波长。
InterSustain C18色谱柱进行了端基封尾,惰性较高,可改善离子型化合物的峰型,而且,由于是改性硅胶基体,pH使用范围(1~10)较广,可用100%水作为流动相,有助于分离极性和亲水性化合物,且更适于选择含无机盐缓冲溶液的流动相系统。本研究选择该色谱柱,能较好地满足实验需求。
消旋山莨菪碱需在UV短波长下进行高灵敏度分析,为避免干扰,应选择不含有机酸类的缓冲溶液。参考《中国药典(2020年版 二部)》和相关文献[6-10],选择甲醇和0.02 M磷酸二氢钾溶液(磷酸调pH值至3.0)为流动相,双嘧达莫、盐酸达克罗宁、消旋山莨菪碱均能得到较好分离。
在本研究中,试验过双波长梯度洗脱法,设置不同时间段流动相的比例,可同时将3种成分进行分离,但是梯度洗脱耗费的时间较长,整个分离过程超过50 min,且四个主要色谱峰的峰形较宽、对称性差,最终采用本文中的方法,简单、快速、准确,实际操作性更强。
消旋山莨菪碱是左旋体和右旋体的混合物。建立的含量测定方法使左旋山莨菪碱和右旋山莨菪碱达到了完全分离,分离度大于1.5。两个同分异构体分别在13、19 min出峰。含量计算参照《中国药典(2020年版 二部)》消旋山莨菪碱片含量测定项下方法,按外标法以消旋山莨菪碱左旋体和右旋体峰面积的和进行计算,准确度和精密度均良好。
综上所述,采用HPLC法测定双嘧达莫乳膏中双嘧达莫、盐酸达克罗宁和消旋山莨菪碱的含量,分离度好,准确度高,对其质量控制和药效评价具有重要意义。
猜你喜欢
西南国防医药(2021年3期)2021-05-14 03:02:08
天津医科大学学报(2021年1期)2021-01-26 00:57:12
中国药业(2020年10期)2020-06-16 08:23:06
文萃报·周二版(2019年24期)2019-09-10 07:22:44
宇航学报(2018年10期)2018-11-08 03:42:54
上海航天(2018年3期)2018-06-25 03:10:34
中国现代医生(2017年28期)2017-11-17 20:03:45
中国现代药物应用(2017年5期)2017-04-06 03:22:12
合成化学(2016年12期)2016-12-27 05:13:29
中国药业(2014年12期)2014-06-06 02:17:45
