
对甲苯基三苯乙炔基硅烷的合成及其固化动力学研究①
2022-09-07 02:50:32唐晓敏曾佳欣李舒平刘美怡谭德新
固体火箭技术 2022年4期
唐晓敏,曾佳欣,李舒平,刘美怡,谭德新
(岭南师范学院 化学化工学院,湛江 524048)
0 引言
含硅芳炔树脂是由芳炔类单体或聚合物经过固化而制备的一类高性能热固性树脂,具备优良的光敏、高残炭率、耐高温等特性,被广泛用于电子、机械、航天航空等领域。目前,国内外学者对芳炔单体合成及其聚合物性能进行了诸多研究。关于芳炔单体的合成主要有两种方法,一种为格利雅试剂(或炔基锂试剂)与氯硅烷反应的传统方法;第二种为多步复杂的其他反应方法。后者较常用的是偶合脱氢反应法,以炔基化合物和氢硅烷为原料,金属化合物( 如RhClL(L=PPh,bp=7,8-benzoquinolinato)、[IrH(HO) (bp)L)]SbF、HPtCl/LiI,IrH(SiEt) (COD) (AsPh)) 为催化剂进行偶合脱氢。由于格利雅反应条件温和,原料廉价易得,操作简单,应用极为广泛,本课题组通过Grignard 反应已制备出系列含硅芳炔单体。此类单体含有刚性芳环结构,交联密度大,在热引发中聚合形成网络状结构,由于在固化反应中无小分子气体溢出,表现出具有优良的成型工艺性能。
固化动力学分析是探究和分析热固性树脂热性能和力学性能的重要中间手段,其研究结果有利于构筑树脂结构与性能关系。目前,研究固化反应主要有红外光谱法、拉曼光谱法、DSC分析法等,而DSC法能够更为直观地获得相关动力学参数,如表观活化能、指前因子和反应级数,同时还能适时获得单体固化程度、固化速率与时间、温度关系。目前,关于芳炔单体的固化行为研究主要集中于非等温固化动力学分析,对芳炔单体的等温固化分析还未见文献报道。
本文以对甲苯基三氯硅烷和苯乙炔为原料,制备了对甲苯基三苯乙炔基硅烷,试图通过改变热聚合单体结构,进一步提高树脂耐热稳定性,并通过热分析方法,对单体固化行为及聚合机理进行了讨论分析。
1 实验
1.1 原料
对甲苯基三氯硅烷(AR),上海吉来德新材料有限公司;四氢呋喃(THF)(AR)、溴乙烷(AR)、无水乙醇、盐酸、甲苯(AR),上海阿拉丁试剂有限公司;苯乙炔(CR),山东淄博汉王公司;镁条,成都市科龙化工试剂厂。
1.2 p-TTPES的合成
将镁条(6.0 g)和THF(50 ml)加入到装有温度计、恒压漏斗、搅拌器和冷凝器的500 ml三颈烧瓶中。向烧瓶中滴加溴乙烷(19 ml)和THF(50 ml)的混合物,加热,保持反应温度在25~40 ℃的情形下回流 3 h。当反应物被冷却到约20℃时,在冰水浴中缓缓加入苯乙炔(27 ml)和THF(50 ml)混合物,加热,在25~30 ℃的条件下回流3 h。当混合物温度降低至室温时,在冰水浴中缓慢加入对甲苯基三氯硅烷(16 ml)和THF(50 ml)混合物,加热,在25~30 ℃下回流3 h。待反应物冷却至室温后,加入盐酸(60 ml,1 mol/L),避免溶液温度升至40 ℃。之后,再加入甲苯(40 ml)进行萃取,通过洗涤分液、干燥、减压蒸馏后,获取黄色油状的粗产物。在低温下加入适量的无水乙醇进行多次重结晶,抽滤后得到DEPES白色粉末状,产率 73 %。反应路线如图1所示。

图1 p-TTPES的反应路线Fig.1 Reaction route of p-TTPES
1.3 测试与表征
(1)傅里叶变换红外光谱测定。采用溴化钾(KBr)压片法,在傅里叶变换红外光谱(美国Nicolet 380型)上进行测试。
(2)核磁共振谱测定。利用超导脉冲傅里叶变换核磁共振波谱仪(德国AVANCE AV-400)进行分析(H-NMR 400 MHz,C-NMR 100.61 MHz,氘代氯仿为溶剂,内标为TMS)。
(3)差热分析测定。使用分析仪(美国Q2000)进行测定。等温DSC测试以50 ℃/min速率升温至指定温度后开始恒温固化,扫描温度为360、365、370、375、380 ℃,测试均在流量为50 ml/min的高氮氛围进行;动态DSC测试则选择5、10、15、20、25 ℃/min的升温速率,升温范围为室温~450 ℃,流量为50 ml/min。
2 结果与讨论
2.1 p-TTPES的结构分析

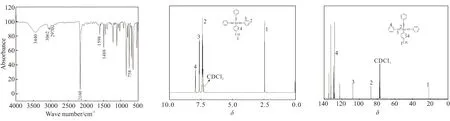
(a) FT-IR (b) 1H-NMR (c) 13C-NMR图2 p-TTPES的FT-IR、1H-NMR 和13C-NMR图Fig.2 Spectra of FT-IR,1H-NMR and 13C-NMR of p-TTPES

通过以上分析,可以确定该合成物质为单体p-TTPES。
2.2 单体等温DSC分析
众所周知,树脂的物化性能通常由其交联网络结构、固化程度以及固化反应的温度和时间决定,而相关动力学参数可提供芳炔树脂结构、性能及加工关系。本文采用等温固化动力学,明确了单体的固化参数,并借助非等温分析对表观活化能进行了验证。通常固化反应根据历程,可分为级模型和自催化反应模型,方程分别见式(1)、式(2):
dd=(1-)
(1)
dd=(1-)
(2)
式中为反应转化率;和为反应级数;、为反应速率常数。
级反应特征是开始时反应速率最大,随时间增长反应速率降低;自催化反应特征为反应具有诱导期,在反应一段时间后速率到达最大值。
由图3(a)可知,反应转化率随固化时间增加而升高;温度越高,固化树脂放热峰值越大,在短时间内可以达到相对较高的转化率。从图3(b)可看出,在反应初期,固化速率随时间增长而快速升高,之后减速,最终趋向平稳。p-TTPES最大固化速率产生在转化率为20%左右,这是自催化反应的典型特征,在诱导期过后,大量的反应位点浓度和体系低粘度有助于固化反应的进行,降低反应位点浓度和增加体系粘度都会显著减慢反应速度。因此,在一定时刻内,固化速率先随固化程度的增加而缓慢增加,到达最大值后,随固化程度增加而降低,最终趋向0。
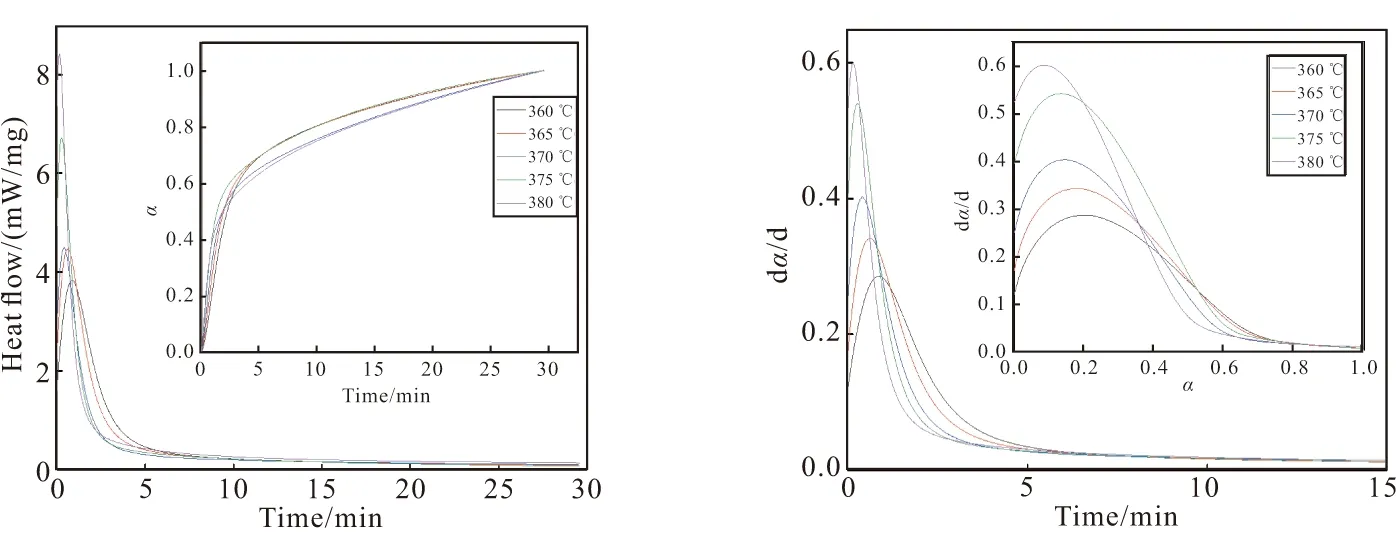
(a) DSC curves (b) (dα/dt)-t curves图3 p-TTPES的等温DSC曲线和(dα/dt)-t关系曲线Fig.3 Isothermal DSC curves and (dα/dt)-t curves of p-TTPES
由图3(b)还可看出,固化反应的初始速率并不为零,速率常数可直接从等温反应速率曲线外推至时间等于零时确定,见表1,同时由公式:
dd=(+)(1-)
(3)
ln(dd)=ln(+)+ln(1-)
(4)
[dd]=0=
(5)
以ln(d/d)对ln(1-)作图,得到图4(a)。

(a) ln (dα/dt) vs ln(1-α) (b) ln{[(dα/dt)/(1-α)n]-k1} vs lnα图4 ln(dα/dt)对ln(1-α)作图和ln{[(dα/dt)/(1-α)n]-k1}对lnα作图Fig.4 Curves of ln(dα/dt) vs ln(1-α) and ln{[(dα/dt)/(1-α)n]-k1} vs lnα
反应级数即为曲线斜率,同时式(4)可重排为
ln{[(dd)(1-)]-}=ln+ln(6)
ln[(dd)(+)]=ln(1-)
(7)
在方程(6)中代入和值,作出图4(b),根据斜率和截距,即可算出反应级数和动力学常数。
为了进一步证实计算方法的准确性,使用上述方程获得第一组参数值;将算出的和值代入方程(7),得到新的反应级数,通过不断重复交互过程,直到所获的各个、和值之间差值低于1 %,最终获得、和值,见表1。
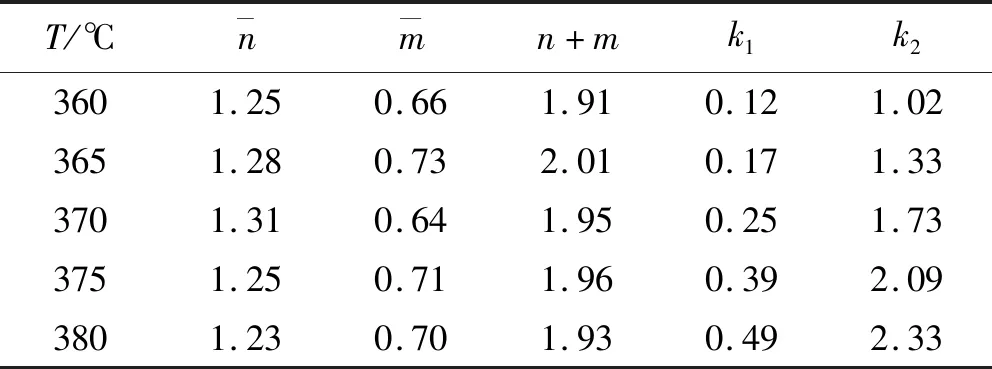
表1 四个动力学参数的值Table 1 Values of the four kinetic parameters
经过计算发现,当+的值约为2时,求得的数据误差最小,在1%以下。由于和与温度满足Arrhenius方程,图5(a)显示ln与1000/呈现很好的线性相关性,以为基础计算的平均活化能为 262.72 kJ/mol,以为基础计算的平均活化能为153.89 kJ/mol。反应起始点的值存在波动,这种波动被认为是导致偏离阿伦尼乌斯关系的原因。因此,通过对ln函数进行线性拟合,得到=153.89 kJ/mol,=5.25×10s,相应的固化动力学模型为
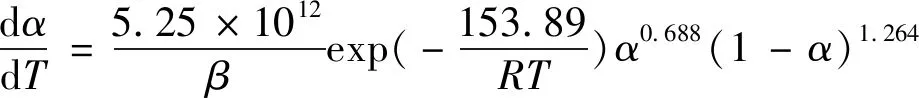
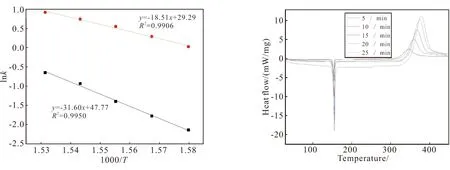
(a) lnk vs 1000/T (b) DSC curves图5 lnk-1000/T作图和不同升温速率下的动态DSC曲线Fig.5 Curves of lnk-1000/T and dynamic DSC curves at different heating rates
2.3 单体非等温DSC分析
p-TTPES的非等温DSC曲线如图5(b)所示,固化反应的起始温度()、峰值温度()、结束温度()及相关参数见表2。随着升温速率增大,固化反应时间由28.5 min缩短到5.3 min,相应固化反应放热峰值向高温方向移动。
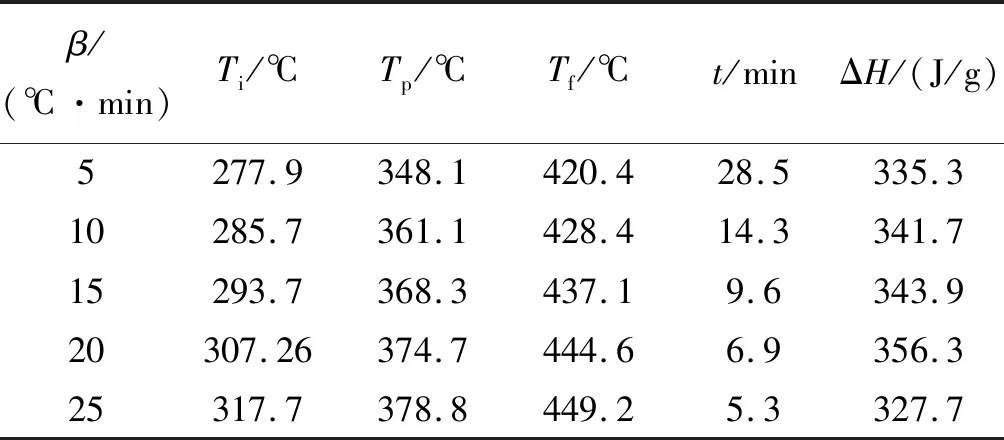
表2 p-TTPES的固化动力学参数Table 2 Curing kinetics parameters of p-TTPES
本文采用Kissinger法、Ozawa法和Flynn-Wall-Ozawa法来计算相关动力学参数,其动力学方程依次如下:
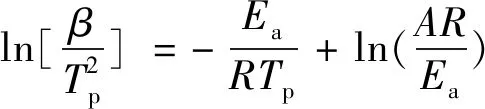
(8)

(9)
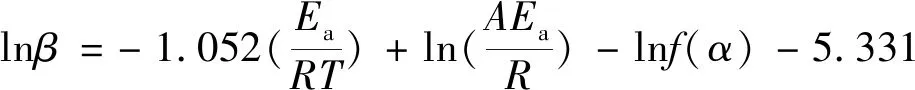
(10)
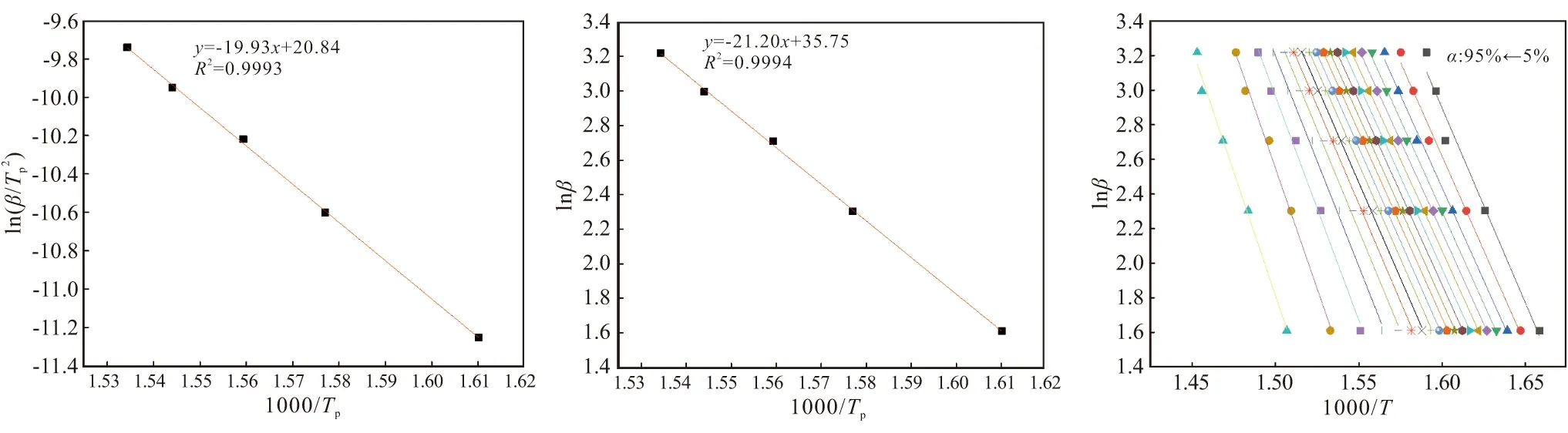
参考文献[14]获得方程拟合图6和相关动力学参数,见表3。从图6可知,曲线相关性较好;表3中,显示三种方法求得反应活化能为166.39 kJ/mol。

表3 三种方法计算的活化能及其相关参数Table 3 Activation energies calculated by three methods and their related parameters
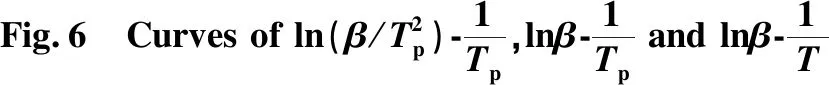
2.4 p-TTPES的固化动力学模型
关于固化反应动力学模型,本文借助Friendman-Reich-Levi法来判断,其表达式为
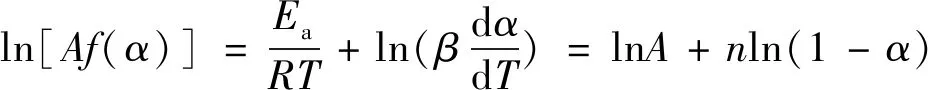
(11)
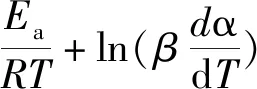
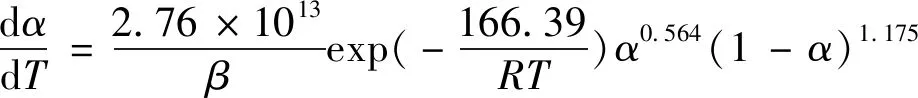
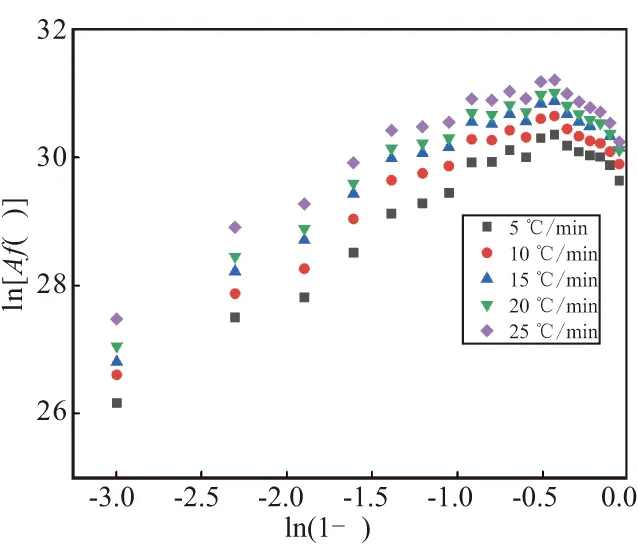
图7 ln[Af(α)]-ln(1-α)关系曲线Fig.7 Curves of ln[Af(α)] -ln(1-α)
通过等温和非等温固化动力学分析可以看出,两种模型计算所得活化能和指前因子相差不大,分别为153.89、166.39 kJ/mol和5.25×10、2.76×10s,这进一步证实实验数据的可靠性。同时,通过与苯基三苯乙炔基硅烷和正己基三苯乙炔基硅烷的非等温固化行为对比发现,其指前因子和活化能相差不大,三者的反应活化能分别为166.39、153.89、158.30 kJ/mol,指前因子分别为2.76×10、9.42×10、6.10×10s。数据分析表明,当芳炔单体取代基碳原子数相近或相同时,结构变化对单体反应的固化行为影响很小,而取代基相差较大时,对树脂性能却有较大影响,主要原因是体系的空间位阻效应较明显,这也进一步说明对于三苯乙炔类芳炔单体,发生热聚合反应时,反应位点浓度较大,增加自由基基元反应的碰撞几率,相似单体的指前因子都较大,反应放热,进一步证实反应为自催化反应。但取代基结构对热稳定性有一定影响,聚正己基三苯乙炔基硅烷和聚苯基三苯乙炔基硅烷树脂在800 ℃下的残炭率分别为60%、73%,而聚对甲苯基三苯乙炔基硅烷树脂最大降解温度为533 ℃,氮气中5%热失重温度()超过460 ℃,800 ℃下残炭率为74%(见图8(a))。可见,芳环及其衍生物的引入对树脂热稳定性有较大影响。为此,需进一步探讨该单体的可能固化机理。
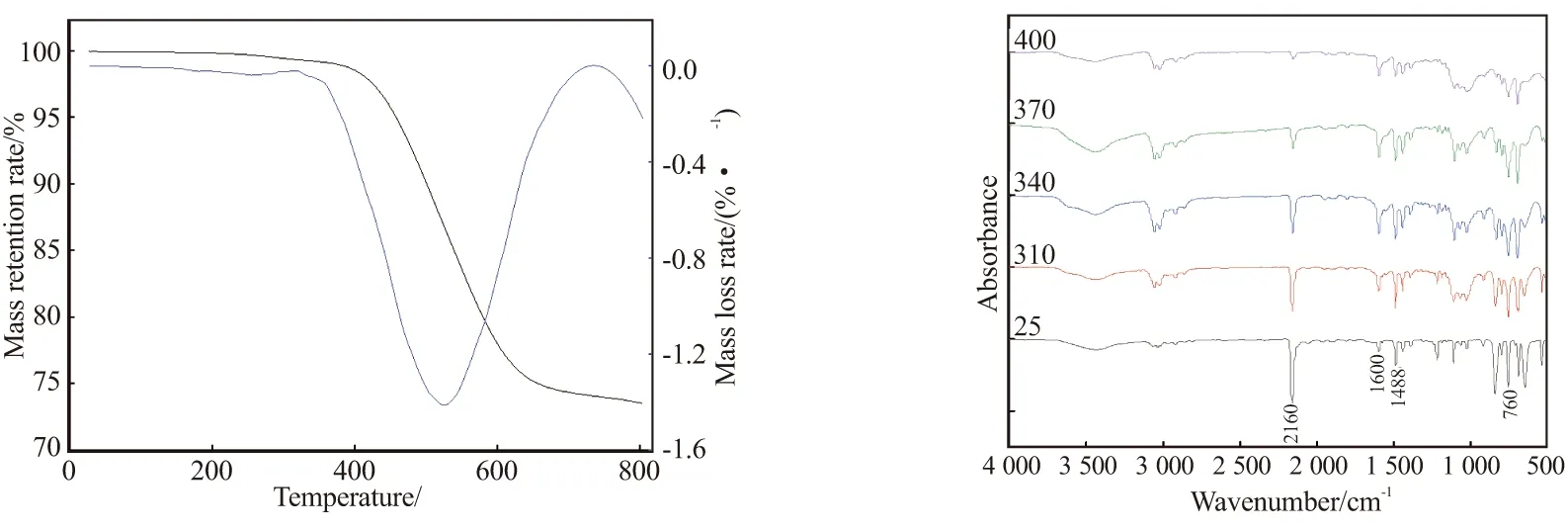
(a) TG curves of polymer (b) FT-IR curves of p-TTPES图8 聚合物热重曲线和p-TTPES在不同温度下FT-IR曲线Fig.8 TG curves of polymers and FT-IR curves of p-TTPES at different temperatures
2.5 p-TTPES的固化分子机理分析

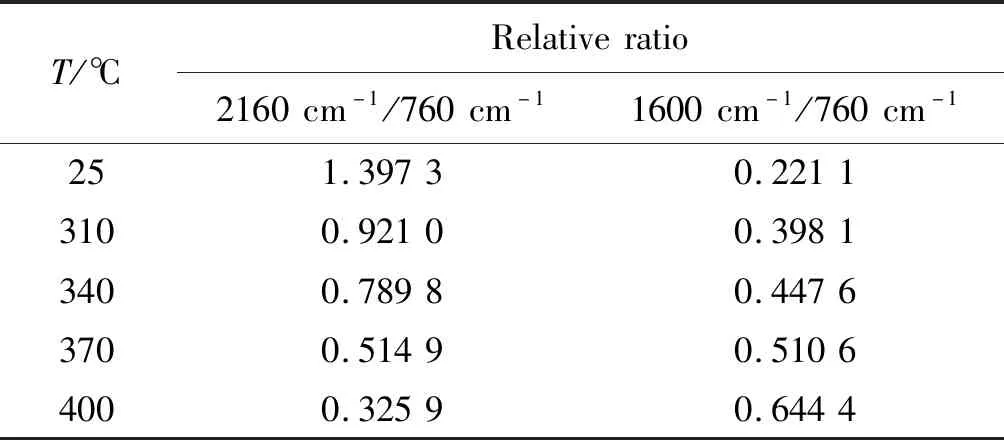
表4 特征峰与参比峰面积的相对比Table 4 Relative ratio of characteristic peak area to reference peak area
根据文献报道,芳炔三键参加热聚合反应主要发生Diels-Alder、环三聚和自由基聚合反应。非等温DSC计算得到固化反应热为341.0 J/g(142.8 kJ/mol),即每个乙炔基的Δ值为47.6 kJ/mol,而RATTO等根据键能计算出发生环化反应的每个乙炔基的Δ值为(189±10) kJ/mol,单体p-TTPES的聚合热远小于预测的环三聚反应的焓变值,这表明乙炔基的环三聚反应不是首选的反应途径,至少在聚合初始阶段不是。从空间位阻来分析,乙炔侧基苯环限制了分子的移动和取向的发生,形成六取代苯环的可能性不大,反应受空间位阻影响,乙炔侧基苯环部分会阻碍三聚反应的必要取向,因此很难发生Diels-Alder反应。从图8(b)中可观察出,波数为1488 cm处苯环吸收峰的变化不大。因此,推测聚合初始状态下形成的产物可能是共轭多烯结构(见图9)。随后,可经历各种加成反应,以产生交联聚合物。

图9 p-TTPES的固化反应机理示意图Fig.9 Diagram of curing reaction mechanism of p-TTPES
3 结论
(1)以对甲苯基三氯硅烷、溴乙烷与苯乙炔为原料进行Grignard 反应,成功制备对甲苯基三苯乙炔基硅烷(p-TTPES)。
(2)等温DSC分析表明,p-TTPES的表观活化能=153.89 kJ/mol,固化反应级数+约为2.0,指前因子=5.25×10s,反应与自催化模型相符。非等温DSC分析获得单体的活化能分别为165.70、167.54、165.94 kJ/mol,两种方法计算出的活化能相近。
(3)热重分析显示,树脂最大降解温度为533 ℃,5%热失重温度()超过460 ℃,800 ℃下树脂的残炭率为74%,显示出优异的耐热耐高温性能。
(4)结合DSC分析和TG分析表明,芳炔单体取代基碳原子数相近或相同时,结构变化对单体反应活化能影响很小,对树脂热稳定性影响较大,从而为该类树脂的合成和工艺化提供参考理论。
