
Angelman综合征的临床及脑电图特征
2022-09-05 06:27陈新卢孝鹏
临床神经病学杂志 2022年4期
陈新,卢孝鹏
Angelman综合征(AS)是母源性泛素蛋白连接酶E3(UBE3A)基因功能异常导致的神经发育障碍性疾病,为较早被发现的单基因遗传性疾病,发病率约为1/150000[1]。近年来各国研究者就AS的发病机制进行了更深入的研究,对不同基因型形成原因、临床表现差异以及再生育风险有了更全面的理解,同时也证实了特征性EEG改变对AS早期诊断具有的重要提示作用[2],是诊断AS的一项敏感指标。本文对我院近年来经基因确诊AS患儿资料进行回顾性分析,探讨不同基因缺陷分型的AS患儿临床及EEG特点,以提高对AS的早期认识。
1 临床资料
1.1 一般资料 系2016—2021年我院经遗传学确诊,有完整视频EEG(VEEG)资料的AS患儿11例。男6例,女5例;年龄10个月至2岁9个月,其中<1岁1例,1~2岁6例,>2岁4例。患儿均足月出生,出生史无明显异常,围产期除1例轻度窒息及1例黄疸外,无特殊异常。否认家族性遗传病史。
1.2 临床表现 见表1。11例患儿均有不同程度的全面性精神运动发育迟缓,特别是运动、语言发育明显落后于同龄儿童。10例患儿皮肤白、眼距宽,5例患儿伴有下颌大、嘴宽、鼻梁低平、牙齿稀疏、伸舌等,4例患儿抽搐与发热相关(1例高热惊厥,3例发热引发抽搐),3例平枕,3例常有与环境不相适应的大笑或微笑,3例入睡困难或睡眠少。
1.3 影像学检查 见表1。所有患儿行头颅MRI检查,其中3例正常,4例脑外间隙稍宽、脑室饱满,2例髓鞘化延迟,1例脑室旁终末带信号偏高,1例脉络膜裂囊肿。
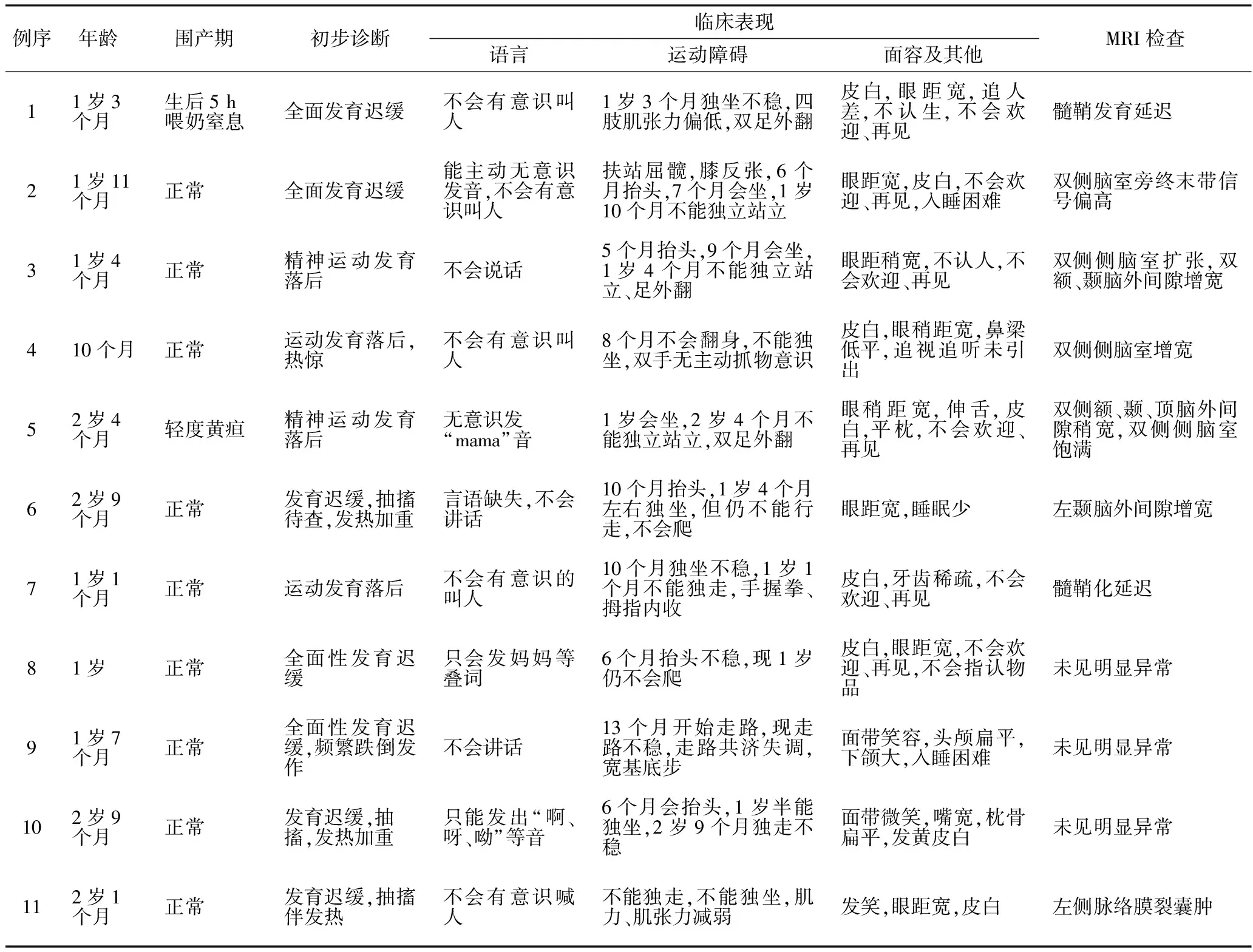
表1 AS患儿临床及影像检查结果
1.4 VEEG检查 见表2。11例患儿共进行21次VEEG监测,均异常,表现为弥散θ波或δ波活动,额、枕区为主δ波活动夹杂棘-慢波,广泛性2~3 Hz棘-慢波/慢波,同一患儿清醒期及睡眠期VEEG未见明显差异。共监测到6例9次不典型失神发作(2次为不典型失神持续状态或伴成串痉挛发作),其中3例(例9、例10、例11)有抽搐病史(全面强直-阵挛、失张力、失神、痉挛发作),另外3例(例2、例5、例7)既往未观察到临床发作。

表2 AS患儿VEEG检查结果
8例片段缺失型及2例移码突变型患儿首次VEEG检查发现,除1例为2~3 Hz δ波背景外,其余9例为弥散4~6 Hz θ波背景;9例监测到局灶棘慢波;10例均表现为额、枕区为主的δ波及广泛性2~3 Hz棘慢波/慢波。1例(例3)父源性单亲二倍体(UPD)患儿首次VEEG改变仅为弥散性θ波,后期复查EEG背景基本正常,伴少量枕区尖慢波、广泛性慢波,较其他分型VEEG异常程度轻。
1.5 遗传学检测 见表3。本组均行遗传学检测,8例(72.7%)为染色体15q11.2-q13.1区域缺失,缺失片段大小为4.8~6.2 Mb;1例(9.1%)为15号染色体为UPD;2例(18.2%)为UBE3A基因点突变(均为移码突变)。

表3 遗传学检测结果
1.6 治疗及预后 6例患儿服用抗癫痫药物,3例为单药治疗,3例为多药治疗。2例(100%)移码突变的患儿均为多药治疗;8例染色体片段缺失患儿中,1例(12.5%)服用2种抗癫痫药物,3例(37.5%)为单药治疗;1例父源性单亲二体患儿未出现抽搐,未服抗癫痫药。5例(例1、例3、例5、例8、例11)患儿随访6个月至3年,无论发作与否,发作间期VEEG均符合AS特征性异常改变,且病程中特征性VEEG异常无明显变化,其中例11在3年内复查7次VEEG,除局灶棘慢波于旁中线区前后变化外,VEEG特征性异常稳定而持续存在。
2 讨 论
AS最早在1965年由英国儿科医生Harry Angelman报道,临床表现包括小头畸形、巨大下颌、张口吐舌、频繁大笑、癫痫、睡眠周期紊乱、肌张力低下、共济失调等[2]。本组患儿语言发育落后突出,其中完全没有语言发育3例,无意识发音7例,仅单音节叠词1例。沈金梅等[3]发现,该组103例患儿均有全面精神运动发育落后,特别是言语发育明显落后于同龄健康儿童。另一组患儿经Gesell发育测评显示,70%中重度发育迟缓,多数为语言重度迟缓[4]。对确诊AS患儿的1~3年随访研究[5]发现,数年后患儿的运动水平和社会交往能力与自身比较均有提高,但语言能力提高不明显。本组11例患儿均存在不同程度的运动发育迟滞。研究[6]显示,UBE3A基因表达异常或缺陷可能降低小脑蒲肯野细胞功能活性,导致小脑性共济失调,引起或加重了患儿的运动功能障碍。本组11例患儿中,10例皮肤白、眼距宽,5例患儿伴有下颌大、嘴宽、鼻梁低平、牙齿稀疏、伸舌等,3例平枕,3例常有与环境不相适应的大笑或微笑。这些症状与AS更为关联,能够辅助AS诊断。AS患儿睡眠障碍及异常的睡眠-觉醒周期在AS诊断标准的共识指南中被提及,本组3例(27.3%)有入睡困难或睡眠少。研究[7]显示,多个多导睡眠图参数表明AS患儿夜间睡眠质量和睡眠效率下降。近年来研究[8]发现,约50%的AS患儿睡眠EEG发现睡眠纺锤波长度显著缩短。这种在非快眼动睡眠期的睡眠纺锤波活动与记忆巩固有关,提示AS患者睡眠质量差和记忆困难存在潜在的直接关系。AS癫痫发生率较高。有报道[3],77.7%的AS患儿伴有癫痫发作,其中80.8%的患儿在3岁以前出现癫痫发作。本组21次VEEG监测中,6例(54.5%)监测到9次不典型失神发作,其中2次不典型失神持续状态或伴痉挛发作。一项纳入43例AS伴癫痫发作患儿的研究[9]显示,失神发作及不典型失神发作占73.5%。杨志仙等[10]分析8例AS患儿的临床资料发现,半数有不典型失神发作,发作间期VEEG示大量广泛性尖-慢波、慢波阵发是失神发作的电生理基础。本组监测到发作的6例患儿中3例(例2、例5、例7)既往未观察到临床发作,但监测中患儿出现意识减低、动作减少的不典型失神发作。可能是患儿存在智力低下、反应性差、共济失调步态等情况,其突然出现的动作停顿,愣神或肢体不自主抖动、震颤未被家长重视。未能及时处理的长期癫痫发作是引起运动认知倒退的重要原因,VEEG监测不仅可以协助诊断,还可以发现一些被临床忽视的发作,使AS患者癫痫发作得到及时有效的治疗。
研究[11]显示,AS与15q11-q13区段UBE3A基因异常有关。导致AS的遗传学机制[12-13]主要包括:(1)母源性染色体15q11-13片段(含有UBE3A基因的5~7 Mb片段)缺失,约占70%;(2)UBE3A基因点突变或其小片段缺失,约占10%;(3)父源单亲二倍体,即一对15号染色体均来自父系,无母系来源,约占2%~7%;(4)印记中心缺陷,使患者颅内无法表达UBE3A基因,约占3%~5%。我国报道的AS患者约二百三十例[14],诊断年龄4个月至6岁,其中母源染色体缺失型199例(86.5%),UBE3A基因突变型14例(6.1%),UPD 8例(3.5%),印记中心缺陷型9例(3.9%)。本组11例患儿中8例(72.7%)为母源性染色体片段缺失,2例(18.2%)为UBE3A基因点突变,1例(9.1%)为UPD,可见染色体片段缺失为AS主要发病的病因,也与上述报道相同。研究[15]显示,AS点突变患者中多数(60%~70%)为小片段基因的缺失和复制导致基因的移码突变,约25%为错义和无义突变,其余为剪切缺陷、复杂重排等。本组中2例基因点突变的患儿均为移码突变,移码突变为点突变主要类型。
UBE3A基因编码的蛋白被称为E6相关蛋白(E6AP),属于泛素蛋白连接酶E3家族,主要定位表达在早期神经元的细胞核和细胞质中,同时广泛存在于神经元的树突和轴突部位,可以维持神经元的正常功能[16]。UBE3A基因异常可导致海马、黑质区的泛素蛋白连接酶减少甚至消失,从而整体影响神经精神系统[17],可通过多种机制导致AS患者出现运动障碍、共济失调、语言障碍、癫痫发作等临床表现[18]。有报道[19]AS患者的临床表型与基因型有一定关系,携带染色体15q11-q13区域大片段缺失患者的临床表型较其他基因型更严重。缺失片段越大、缺失片段距离着丝点越近,语言损害、运动障碍和癫痫可能越严重。患儿年龄较小时部分症状还未表现出来[20],随着年龄增长,AS相关的表现更加明显。本研究发现,2例(100%)移码突变患儿均为抗癫痫药物多药治疗;8例片段缺失患儿中1例(12.5%)例为多药治疗;1例UPD患儿未服用药物,UPD患者15号染色体均来源于父亲,染色体结构、数量并无异常,癫痫发病率相对较低[18],而UBE3A基因移码突变可能对患儿神经精神系统影响比其他类型突变更严重[21],导致患儿癫痫较难控制。但由于本组病例数量较少,有待更大样本的研究。
由于AS患儿年龄较小,且临床表现缺乏特异性,婴儿期极易被误诊为脑瘫、发育迟缓等,多数AS早期诊断得益于EEG特征性异常[22]。目前较公认的AS患儿的异常EEG改变[10,23]包括:(1)与闭眼无关的持续广泛性或后头部4~6 Hz脑波;(2)前额区高波幅2~3 Hz的慢波、三相δ波,常夹杂中波幅棘波、尖波,慢波活动可趋于泛化全导,常在清醒时即出现,在睡眠状态下波形振幅降低而不会消失;(3)枕区出现3~4 Hz的高波幅棘(尖)波、尖慢波混合高波幅慢波,有时双侧不对称。上述异常可单独或混合出现。本组患儿21次VEEG均异常(100%),具有特征性:发作间期80.9%为弥散性4~6 Hz θ波活动;17次(80.9%)出现额、枕区δ波节律,19次(90.5%)出现广泛性2~3 Hz棘-慢波阵发;80.9%的患儿棘-慢波于多个部位(额、中央、顶、枕区),枕区更突出。研究[24]表明,AS患儿在清醒期及睡眠期都表现出较强的δ振荡,且在整个新皮质中普遍存在,不会因睁闭眼等简单刺激而消失[25],也不会因为睡眠期或清醒期而改变[26]。这个特点可以区分AS患儿与其他严重发育落后伴有或不伴有癫痫发作的患儿[23]。本组患儿无论发作与否,发作间期VEEG均符合AS特征性异常改变,同一次监测中醒睡期VEEG特征未见明显差异,病程中复查VEEG,AS患儿的特征性EEG异常持久而稳定,因此对患儿早期诊断有重要提示作用。但VEEG异常并非特异性,因此运动语言发育严重落后、伴有特征面容的患儿出现特征性EEG异常改变时,需进行基因学检查明确诊断。Frohlich等[27]将缺失基因型、非缺失基因型AS患儿及正常对照组EEG进行比较发现,与非缺失基因型相比,缺失基因型的θ功率(峰值频率5.3 Hz)升高,β功率(峰值频率23 Hz)降低;与正常对照组比较,二者EEG功率在广泛的频率范围内(1~32 Hz)升高,最大差异是δ频段(峰值频率2.8 Hz),且δ功率在患儿在整个发育过程中稳定高于对照组。Sidorov等[24]使用定量分析法对AS患儿的EEG进行研究证实,AS患儿在δ频带中功率值显著增高,因此大量的δ波振荡是最突出的EEG表型,这一结果与本研究是一致的。
综上所述,AS患儿运动、语言发育明显落后于同龄儿童,不典型失神为常见的癫痫发作类型,特征性异常EEG持久且稳定,大量的δ波振荡是最突出的EEG表型,此类电-临床特征对AS患儿早期诊断有重要提示作用。
猜你喜欢
癫痫与神经电生理学杂志(2022年2期)2022-06-07
健康必读·下旬刊(2019年8期)2019-08-16
科学之谜(2019年3期)2019-03-28
科学之谜(2018年8期)2018-09-29
家庭百事通·健康一点通(2017年11期)2017-11-29
饮食科学(2017年5期)2017-05-20
中学生理科应试(2016年4期)2016-11-19
家庭医药(2016年8期)2016-09-28
恋爱婚姻家庭·养生版(2016年9期)2016-09-07
意林(2013年15期)2013-05-14
