
copGFP和PuroR双标记基因慢病毒融合表达载体的构建及应用*
2022-08-29 01:49朱文琦赵思捷李永利卞成蓉鲍春梅李伯安
中国应用生理学杂志 2022年2期
张 浩, 朱文琦, 赵思捷, 李永利, 侯 俊, 卞成蓉, 鲍春梅, 卫 桢, 孙 杰,李 波△ , 李伯安△
(1. 解放军总医院第五医学中心检验科, 2. 解放军总医院第五医学中心感染病研究所, 北京 100039 )
慢病毒过表达系统在基因功能研究、假病毒感染宿主能力测定及药物筛选、CAR-T免疫细胞治疗等过程中发挥重要作用[1-5]。由于慢病毒包膜蛋白为来自于水疱疹病毒的VSV-G蛋白,其受体为细胞膜上表达的磷脂酰丝氨酸,因此其宿主范围广泛,包括分裂和非分裂细胞;另外,由于慢病毒感染细胞后,其RNA经逆转录酶转录、扩增为双链DNA后随机整合入宿主细胞基因组中,可实现目的基因长期稳定表达,因此,慢病毒过表达系统在基础研究、生物治疗、药物筛选模型建立等领域越来越受到青睐[6-8]。
为获得稳定整合慢病毒基因组的细胞,可以通过抗生素抗性基因和绿色荧光蛋白两种筛选标记基因进行筛选得到。抗生素(G418、嘌呤霉素、博来霉素等)一方面可杀死未感染的细胞,另外,给予稳定整合慢病毒基因组的细胞抗生素压力还可以避免整合序列的丢失;绿色荧光蛋白的表达则使感染的细胞通过荧光显微镜定性观察、流式细胞仪定量检测以计算包装慢病毒的滴度、流式细胞仪对阳性细胞的分选成为可能,因此在许多商业化的慢病毒载体中同时含有绿色荧光蛋白和抗生素两个标签,但这两个标签编码序列大都通过IRES或T2A等序列连接,它们经转录后形成的mRNA虽然位于同一条链上,但经翻译后形成两个独立的蛋白质[9,10],不同的蛋白质由于其氨基酸序列不同,它们在细胞内的翻译后修饰不同,其半衰期和实际的表达水平或强度不一定相同,所以在感染细胞中,抗生素抗性基因与绿色荧光蛋白表达水平不完全一致,在抗生素抵抗能力强的细胞中绿色荧光蛋白表达水平表达也不一定就很强,这为后续的分析筛选工作造成干扰。为克服这一潜在的缺陷,本研究运用重组PCR技术将源于桡足动物的copGFP(绿色荧光蛋白)和PuroR(嘌呤霉素抗性基因)编码序列直接融合在一起,并克隆至慢病毒表达载体中,随后将该载体包装成慢病毒后感染肝癌细胞,检测感染细胞对抗生素的抵抗作用和绿色荧光蛋白的表达水平。
Sp1为Sp/KLF转录因子家族成员(该家族已发现26个成员),含有785个氨基酸,研究表明Sp1参与肺癌、卵巢癌、结直肠癌等肿瘤细胞增殖、存活、迁徙和转移、炎症反应和药物抗性等过程[11,12],为研究该分子在肝癌发生发展过程中的意义,将较长的2359 bp的Sp1编码片段克隆至改构的融合表达双标签载体中与空载体比较检测其在肝癌细胞中的表达水平以确定该载体在基因过表达中的效能以及为Sp1在肝癌发生发展中的作用研究奠定基础。
1 材料与方法
1.1 材料
人肝癌细胞系MHCC97H、人胚胎肾细胞293T、pLKO.1、pCDNA-FLAG-Sp1载体及慢病毒包装质粒, pCDH-CMV-MCS-EF1-copGFP载体由本中心保存;PyrobestTMDNA聚合酶、T4 DNA连接酶、Sal I和BamH I限制性内切酶(大连宝生物公司),胎牛血清(杭州四季青公司),DMEM(Hyclone公司),嘌呤霉素(Amresco公司);PCR产物回收试剂盒、胶回收试剂盒、质粒提取试剂盒为天根生化有限公司产品,反转录回收试剂盒、定量PCR反应试剂盒为Takara公司产品、Sp1抗体及b-actin抗体为美国CST公司产品;HRP标记的鼠抗兔二抗为美国Jackson实验室产品;蛋白酶抑制剂cocktail为罗氏公司产品;Western blot发光液由普利莱公司提供;本研究所用引物由北京博迈德基因技术有限公司合成。
1.2 pCDH-CMV-MCS-EF1-copGFPuro双标签融合表达载体的构建
⑴ 从pCDH-CMV-MCS-EF1-copGFP载体中扩增出多克隆位点BamH I至copGFP终止密码子前的1 340 bp片段,PCR扩增所用的上游引物为:5’-CCGGAATTCGAATTTAAATCGGATCC-3’ (划线部分为限制性内切酶BamHI识别位点),下游引物:5’-GTGGGCTTGTACTCGGTCATGCGAGATCCGGTG-GAGCC;⑵ 以pLKO.1质粒为模板扩增670 bp的puromycin编码序列,PCR扩增所用的上游引物为:5’-ATGACCGAGTACAAGCCCAC-3’;下游引物:5’- ACGCACGCGTCGACTCAGGCACCGGGCTTGCGGG-3’ (划线部分为限制性内切酶SalI识别位点);⑶以该产物为模板,在5’端重叠延伸出与copGFP编码区末端18个碱基序列互补的PCR扩增产物,上游引物序列为:5’-GGCTCCACCGGATCTCGCATGACCGAGTACAAGCCCAC-3’(划线部分为重叠延伸区域);下游引物:同上一步;⑷ 以第⑴、⑶步PCR为模板,重组延伸出含copGFP与PuroR编码序列融合表达的片段,上游引物同第一步上游引物,下游引物序列同第三步下游引物。重组PCR产物经BamH I+Sal I双酶切,与经同样双酶切的pCDH-CMV-MCS-EF1-copGFP片段连接、转化后,挑取单克隆进行测序确证,构建过程参见图1。
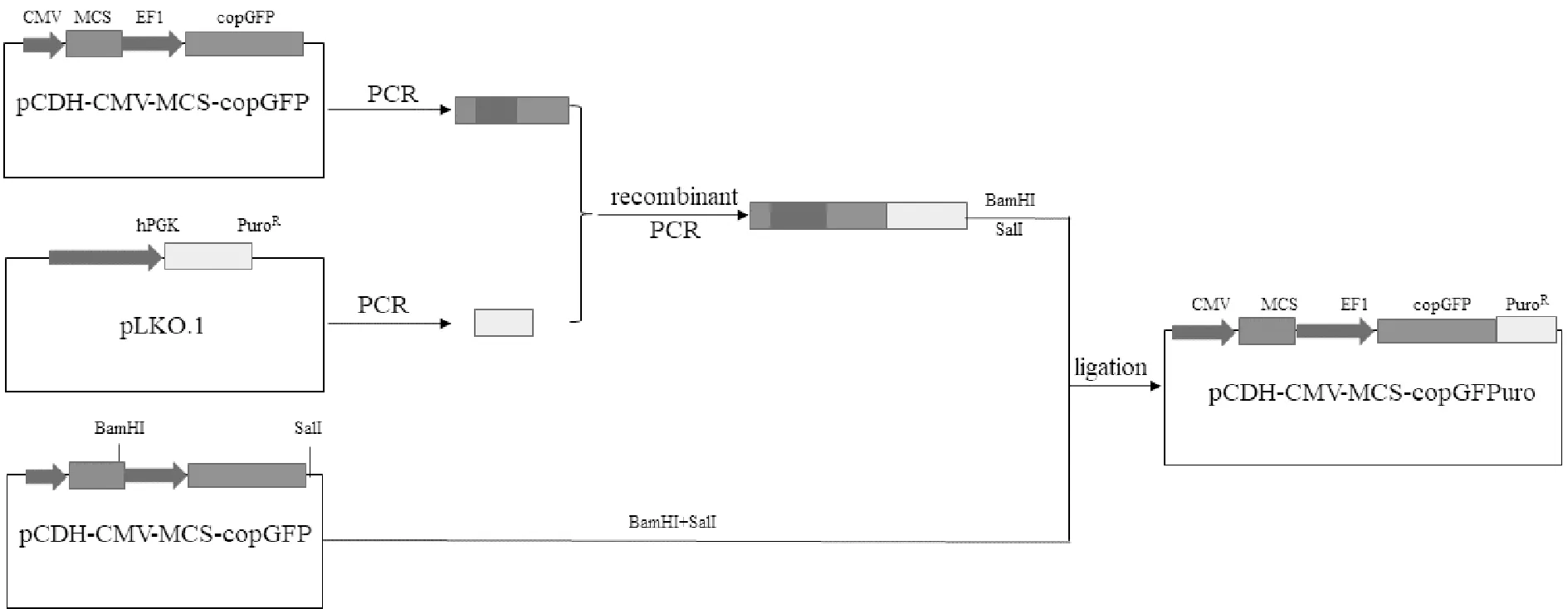
Fig. 1 Schematic representation of the construction of pCDH-CMV-MCS-EF1-copGFPuro vector
1.3 pCDH-CMV-Sp1-EF1-copGFPuro载体的构建
首先从pCDNA-FLAG-Sp1载体中扩增出FLAG-Sp1编码区段,上游引物为:5’- GCTCTAGAGCCACCATGGACTACAAGGACGACGA-3’,下游引物:5’-CCGGAATTCTCAGAAGCCATTGCCACTG-3’ (划线部分为限制性内切酶识别序列),PCR产物经Xba I+EcoR I双酶切,与经同样双酶切的pCDH-CMV-MCS-EF1-copGFPuro片段连接、转化后,菌液PCR筛选阳性克隆,提取质粒进行双酶切分析和CMV测序引物进行测序确证。
1.4 慢病毒的包装及感染及puromycin筛选
参考文献[13] ,在包装含Sp1编码序列的慢病毒载体时,由于Sp1编码序列较长(2 358 bp),在转染293T细胞时,将转染含Sp1编码序列的慢病毒载体的质量提高1倍。即在6孔板中,转染PLP1、PLP2、VSVG与表达Sp1慢病毒载体的量分别为1 mg、0.5 mg、0.7 mg和2.5 mg;转染后4 h换液,24与48 h后分别取细胞培养上清,4℃ 3 000 r/min离心10 min,取100 ml感染细胞(同时加入8 mg/ml polybrene),其余-80℃保存。感染12 h后,换液;感染24 h后加入嘌呤霉素处理。
细胞感染前,分别加入不同浓度(0.125、0.25、0.5、1、1.5和2 mg/ml)的嘌呤霉素,选择导致细胞7 d死亡的最低浓度为感染慢病毒细胞的筛选最佳浓度,对MHCC97H细胞而言,1 mg/ml嘌呤霉素为最佳筛选浓度。感染细胞时,同时设未感染细胞以及感染含对应过表达载体的慢病毒作为空白对照和阴性对照。
1.5 RT-qPCR检测Sp1 mRNA在肝癌细胞中的表达水平
用Trizol法分别提取稳定表达对照载体以及稳定表达Sp1表达载体的MHCC97H细胞的总RNA,取0.5 mg反转录为cDNA并稀释5倍后,进行实时荧光定量PCR检测,用于检测Sp1的引物为:正向引物5’- TGGCAGCAGTACCAATGGC-3’,反向引物5’- CCAGGTAGTCCTGTCAGAACTT -3;用于检测b-actin的引物为:正向引物5’-CATGTACGTTGCTATCCAGGC-3,反向引物5’-CTCCTTAATGTCACGCACGAT-3,通过各自的CT值计算出HBX敲低后其mRNA在细胞内的相对表达量。
1.6 Western blot检测Sp1蛋白在肝癌细胞中的表达水平
慢病毒上清感染肝癌细胞MHCC97H经嘌呤霉素筛选一周后,传代至6孔板中继续培养,待细胞铺满80%后,传代至新的6孔板中,细胞铺满80%后,用预冷的1 ml 1×PBS洗一次,再加入700 ml PBS,用细胞刮子将细胞刮下,10 000 r/min离心20 s,弃上清,加入100 ml RIPA裂解液,用200 ml加样器枪头轻轻将细胞搅匀,冰浴10 min,超声破碎仪破碎(比利时,Diagenode),4℃ 12 000 r/min离心10 min,取上清,BCA法进行蛋白定量,取20 mg 进行10% SDS-PAGE蛋白电泳,半干法转印蛋白至0.45 mm硝酸纤维素膜上,分别用Sp1(1∶500)和β-actin(1∶10 000)孵育,再加入HRP标记的二抗反应1 h后,在发光液中反应1 min,在全自动化学发光成像分析系统(上海天能科技有限公司)中进行条带分析。
2 结果
2.1 copGFP和puromycin融合表达双筛选标记慢病毒过载体的构建
分别从pCDH-CMV-MCS-EF1-copGFP和pLKO.1载体中扩增出1 300 bp含copGFP编码序列以及670 bp PuroR编码序列的DNA片段,经重组PCR后,得到1 970 bp的重组序列,见图2A第1、2、3道;重组序列经BamH I+Sal I双酶切后与用同样酶切的pCDH-CMV-MCS-EF1-copGFP载体片段连接,转化,挑取单克隆细菌培养后提取质粒进行双酶切鉴定,见图2A,第4道为没有进行酶切的pCDH-CMV-MCS-EF1-copGFPuro重组质粒,第5,6道分别为用BamH I+Sal I双酶切的pCDH-CMV-MCS-EF1-copGFP和pCDH-CMV-MCS-EF1-copGFPuro载体,图中可见后者BamH I+Sal I中的插入片段显著大于前者切出的小片段,提示新重构的载体中含有新插入的PuroR编码序列,为进一步验证copGFP和PuroR的融合表达,对重构载体融合编码标签区域进行序列测定,表明序列与预期一致,结果见图3,用反向引物测序后,发现copGFP和PuroR的编码序列融合在一起。

Fig. 2 DNA electrophoresis showing the process of recombinant PCR(A) and double restriction endonuclease analysis constructed vector (B)

Fig. 3 Result of reverse sequencing of the fusion coding region of copGFP and PuroR
2.2 copGFP和puromycin融合表达双筛选标记在肝癌细胞中的表达
分别将只含有copGFP筛选标记的原始载体以及含copGFP和puromycin融合表达载体与辅助在293T细胞中包装慢病毒,肝癌细胞MHCC97H,经1 mg/ml的puromycin筛选7 d后,发现没有感染的对照细胞以及感染只含有copGFP筛选标记的对照组,细胞全部死亡(图4A3和A4),而感染含有copGFP和puroR融合表达的慢病毒实验组细胞开始虽有部分细胞死亡,但经过7 d后,由于感染细胞的持续增殖,细胞孔接近80%铺满(图4A2),在荧光显微镜下观察这些细胞成梭形的伸展细胞,可以明显发现绿色荧光蛋白的表达(图4A1),提示融合的双标签同时具有两个筛选标记基因的功能。为验证copGFP和puromycin的融合表达,提取慢病毒感染细胞总RNA,进行反转录,并分别运用针copGFP编码区 5’和puroR3’端的引物进行PCR反应,结果扩增出1.4 kb大小的片段,与copGFP和puroR融合蛋白编码区大小一致,而在没有感染的对照细胞中没有扩增出相应片段(图5)。

Fig. 4 Microscopic photograph (×200) of MHCC97H cells infected with lentivirus containing copGFP and PuroR fusion marker (A1, A2) or infected with lentivirus containing only copGFP labeling gene (A3) or without infection as blank control (A4), section A1 is photographed in the presence of excitation wave showing the green fluorescence emission from puromycin resistant cells
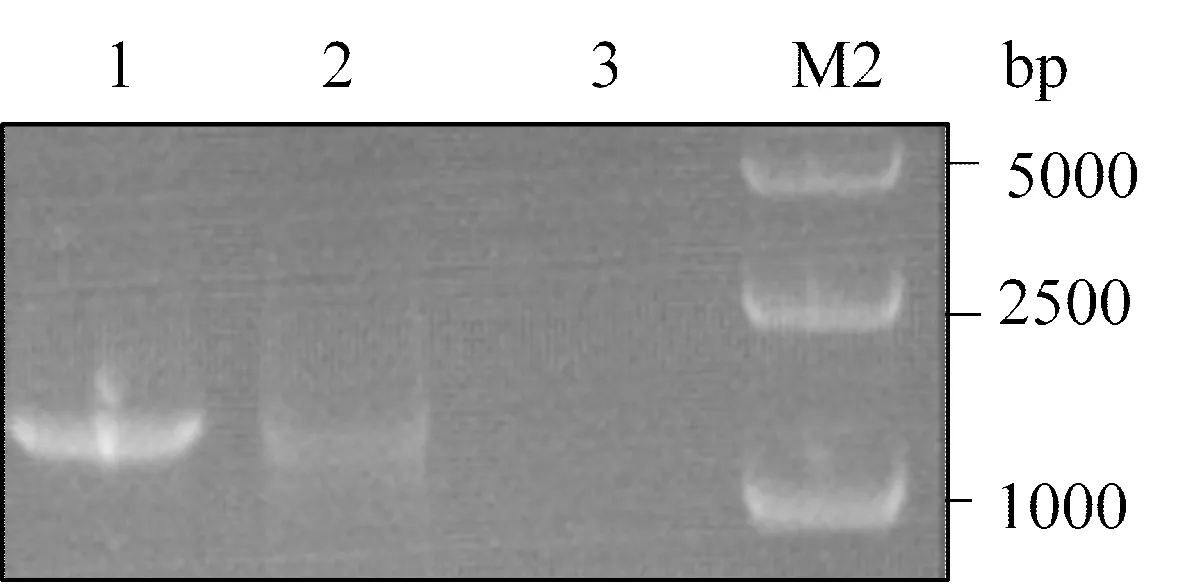
Fig. 5 RT-PCR analysis of copGFP and puroR fusion protein expression in infected MHCC97H cells
2.3 copGFP和puromycin融合双筛选标记的慢病毒过载体启动目的基因表达能力的验证
为验证该载体启动插入目的基因的表达能力,将人转录因子Sp1克隆入该载体中,包装为慢病毒后感染肝癌细胞经嘌呤霉素筛选后MHCC97H获得稳定表达的细胞系,细胞经传代后分别提取总RNA及总蛋白,分别运用RT-qPCR和Western blot方法检测Sp1 mRNA和蛋白表达水平。mRNA结果参见表1,和对照细胞相比,稳定表达载体插入Sp1的细胞系中,Sp1 mRNA水平升高3.3倍;Western blot结果显示,与未感染组对照细胞相比,Sp1蛋白表达水平升高2.2倍(图6,表1)。

Fig. 6 Western blot analysis of protein levels in MHCC97H cells infected with lentivirus packaged with vehicle control vector or with Sp1 coding sequence inserted vector

Tab. 1 Real-time PCR analyses Sp1 mRNA n=3)
3 讨论
带有双筛选标记基因的慢病毒表达载体已广为应用,通常一个标记基因编码抗生素抗性基因,通过抗生素处理杀死未感染细胞,另一个标记基因为绿色荧光蛋白(EGFP),可运用流式细胞术分选出表达阳性的细胞。商业化的慢病毒载体中通常在这两个标记基因之间添加内部核糖体进入位点(IRES)或T2A序列,最终产生两个独立的蛋白质分子,由于蛋白分子存在复杂的翻译后修饰,因而无法保证两个筛选标记基因表达的一致性,这为后续筛选表达不同强度标记基因的细胞带来不确定性[14],因此本研究旨在将两个筛选标记基因在慢病毒载体中进行融合表达,由于两个标记分子融合在同一个蛋白分子中,从保证它们表达时间的同步性和强度的一致性。融合表达的另一优势在于两个筛选标记基因之间没有其它外源插入序列,尽量减少载体的长度,有利于提高慢病毒包装效率;另外,考虑到桡足类生物copGFP荧光强度超过来源于维多利亚多管发光水母(aequorea victoria)EGFP荧光强度的1.3倍,作为筛选标记基因在研究中得到广泛应用[15,16],本实验选用copGFP与嘌呤霉素编码序列进行融合表达;同时经本实验改构后的载体与原始载体PCDH-CMV-MCS-copGFP的多克隆位点一致,原有的限制性酶切位点均可在克隆中方便选择使用。
改构的慢病毒空表达载体包装为病毒后感染肝癌细胞后,经嘌呤霉素筛选7 d后,对照未感染以及感染只表达copGFP标记基因慢病毒的细胞全部死亡,而感染含copGFP与PuroR融合表达双筛选标记基因的细胞则持续增殖至70%铺满,表明copGFP与嘌呤霉素融合表达后具有对嘌呤霉素的抵抗作用;这些经嘌呤霉素筛选后的细胞在荧光显微镜下观察可见到绿色荧光蛋白的表达,提示嘌呤霉素并抗性基因片段不影响copGFP标记基因的功能,在激发光的作用下融合标记基因可发射出绿色荧光。
在本研究中,将2 359 bp的Sp1编码片段克隆至改构的融合表达双标签慢病毒表达载体中,包装成慢病毒后感染肝癌细胞MHCC97H,经RT-qPCR和Western blot鉴定发现在感染编码Sp1编码片段的细胞中,与对照空载体感染组相比,感染含Sp1编码序列的细胞组中,Sp1的mRNA和蛋白表达水平均得到显著提高,表明构建的慢病毒双标签融合表达载体与辅助包装质粒共转染293T细胞后可以包装出含较长外源插入片段的慢病毒,并在真核细胞中进行高效表达;本研究也为揭示Sp1在肝癌发生发展中的作用机制奠定基础。
综上,经改构的含copGFP与嘌呤霉素抗性基因融合表达双筛选标记的慢病毒载体具有表达绿色荧光蛋白和嘌呤霉素抗性的特性,并可表达较长片断的目的基因,该新型双标签融合表达载体将为后续基因功能等相关研究奠定基础。
猜你喜欢
华人时刊(2022年9期)2022-09-06
中国现代医生(2022年21期)2022-08-22
中国糖料(2022年3期)2022-07-04
中国农学通报(2022年12期)2022-06-01
昆明医科大学学报(2022年1期)2022-02-28
作物学报(2022年4期)2022-02-10
广东教育·职教版(2021年3期)2021-04-20
华人时刊(2020年15期)2020-12-14
中学生物学(2019年7期)2019-10-17
人大建设(2018年6期)2018-08-16
