
国槐不同器官转录组分析及EST−SSR引物开发应用
2022-08-26 10:59:16徐立人庞新博刘朝华李泳潭原阳晨
西南林业大学学报 2022年4期
郁 洁 徐立人 庞新博 刘朝华 李泳潭 原阳晨 董 研
(1. 河北农业大学林学院,河北 保定 071000;2. 河北省林木种质资源与森林保护重点实验室,河北 保定 071000;3. 河北农业大学园林与旅游学院,河北 保定 071000;4. 河北省洪崖山国有林场,河北 保定 074200)
国槐(Sophora japonica),又名家槐、中华槐等,为豆科(Leguminosae )槐属多年生落叶乔木。国槐的栽培历史悠久,约在3000年以上,我国古代最早的词典《尔雅》有关于槐的记载,《山海经》中有“首山其木为槐”的记述[1]。国槐原产地为我国北部,常见于华北平原和黄土高原,现全国各地均有栽培,是我国常见的绿化树种。国槐树高可达25 m,树干表皮灰褐色,表面粗糙纵裂,小枝绿色,皮孔明显,花期一般在6—7月,可作为优良的蜜源植物[2]。其萌芽力强、生长快、极耐修剪,在涵养水源、防风固沙、水土保持等环境方面有重要的作用[3]。基于国槐的诸多优良特性,我国的科研工作者进行了长期的国槐新品种选育工作,相继选育出了一批国槐优良品种,如聊红槐、华箭、嫣红3号[4−6]等。
DNA分子标记技术能够精确地进行遗传多样性分析。现已开发出RAPD、SRAP、ISSR、AFLP等多种标记手段,并已应用到多种植物的遗传多样性评价中[7]。SSR(simple sequence repeats)分子标记也称微卫星序列标记,是一类由若干个核苷酸(1~6个)为重复单元串联重复组成的DNA序列,广泛分布在真核生物的基因组中,其较传统的形态标记、生化标记等具有共显性、稳定性高、可重复性强等优点,在研究生物遗传多样性、亲本鉴定、DNA指纹图谱构建和分子标记辅助育种等众多方面发挥着重要作用[8−9]。SSR标记根据来源可分为基因组SSR(G−SSR)和表达序列标签SSR(EST−SSR),G−SSR标记来源于基因组序列,一般采用构建和筛选基因组文库、微卫星富集和数据检索等经典方法进行开发,该类标记极少能定位到基因上。EST−SSR标记来源于转录序列,是生物体某一组织在特定时期的表达基因,可定位在相关的功能基因上,且具有种属间的通用性[10−11]。目前,已有大量关于利用转录组测序开发不同物种EST−SSR分子标记的报道[12−13],国槐作为我国优良的乡土树种,其遗传多样性研究仅限于扩增多态性(SRAP)、简单序列间重复序列(ISSR)及少量表达序列标签(EST−SSR)[14−16]等,EST−SSR引物资源相对较为匮乏。本试验以11个不同国槐无性系为材料,从“青云1号”无性系中随机选择一株,对其根、茎、叶不同器官进行转录组测序,进行GO及 KEGG功能富集,进一步对不同器官中EST−SSR的分布情况进行初步分析,开发出一套EST−SSR标记,进而构建11个国槐无性系的进化树及指纹图谱,以期为今后国槐的种质资源保护、新品种开发及生物信息学研究提供参考依据。
1 材料与方法
1.1 材料来源
试验所在地为河北省洪崖山国有林场管理局七里亭示范林场,位于河北省保定市易县京环线(N 39°35′99″,E 115°56′36″),属温带大陆性季风气候,四季分明,雨热同期。年平均气温12.2 ℃,年降水量570 mm,无霜期 200 d,年平均日照 2578.3 h,太阳辐射量128 J/(m2·d)。
以河北省林科院选育的“青云1号”国槐品种为对照(CK)及2016年在河北省洪崖山国有林场国槐实生林中选出同一种源地内10株树干通直、生长健壮的单株为材料(编号为A−1~A−10),2019年3月,采用完全随机区组试验设计,以2年生普通国槐为砧木,以低接的方式,构建11个不同国槐无性系的对比试验林,设3个区组,每个区组11个小区,每个小区栽6株,株行距为3 m ×4 m。
1.2 研究方法
1.2.1 转录组测序
本试验以2年生国槐品种“青云一号”为材料,随机选取一株生长健壮、无病虫害的国槐无性系,分别从上、中、下3层采集其根、茎、叶不同部位的新鲜组织3次,随后进行混样,液氮速冻后将其放入干冰中,送至北京诺和致源科技股份有限公司进行高通量转录组测序,本研究中各样品的Q30碱基百分比均在93.19%以上,测序质量较高,满足后续试验要求。
1.2.2 DNA提取
2020年7月28日,于每个无性系采集生长健壮、无病虫害的嫩叶,放入装有变色硅胶的PE自封袋中,带回实验室,采用改良的CTAB 法[17]提取各样本的总DNA,将检测合格的DNA统一稀释至20 ng/μL,放入−65 ℃冰箱中保存备用。
1.2.3 EST−SSR引物设计与筛选
采用MISA (http://pgrc.ipk-gatersleben.de/misa/misa.html ) 软件查找转录组数据中的SSR位点。标准为:单、二、三、四、五、六核苷酸重复单元最低重复次数分别为10、6、5、5、5、5次。参考 Guo 等[18]的方法,使用Primer 3软件进行EST−SSR引物的设计与筛选,标准为:1)GC碱基含量为40% ~ 60% ;2)引物长度设置在18~25 bp;3)预期扩增产物大小在100~300 bp;4)退火温度为52~65 ℃,且同一引物的正向引物和反向引物温度差小于0.6 ℃;5)引物之间无引物二聚体结构的出现。进一步从符合筛选标准的引物中随机挑选30对,交由上海生工生物工程股份有限公司合成。
1.2.4 PCR扩增体系及程序
PCR扩增体系采用20 μL体系,各成分及比例 为:2 × M5 HiPer plus Taq HiFi PCR mix(Blue)10 μL,正向引物和反向引物各1 μL (10 ng/μL),DNA模板1 μL (20 ng/μl),最后用去Nuclease-free dd H2O补足至20 μL。
PCR扩增程序为:95 ℃预变性3 min;94 ℃变性25 s,55 ℃退火25 s,72 ℃延伸15 s,33个循环;72 ℃延伸5 min;4 ℃保存。PCR扩增产物用8%非变性聚丙烯酰胺凝胶电泳进行检测,电压设定为230 V,时间为35 min,电泳结束后进行银染显色及拍照记录。
1.2.5 数据统计与处理
利用EXCEL 2016软件对全部Unigene序列中SSR位点出现的频率、分布规律、数量及类型进行统计;将8%非变性聚丙烯凝胶电泳所得到的图片,转换为0/1矩阵,有扩增条带的记为1,反之为0 ;利用 Cervus 3.0.7 软件计算各引物的多态性信息量(PIC);利用 POPGENE 32软件统计具有多态性引物的 Shannon's 指数(I)、等位基因数(Na)和有效等位基因数(Ne)等;利用DPS 7.05 软件,利用类平均法(UPGMA)计算各样本间的遗传距离矩阵并绘制进化树,构建指纹图谱。
2 结果与分析
2.1 国槐不同器官转录组差异分析
2.1.1 测序结果分析
通过转录组测序,对国槐根、茎、叶3个不同器官的转录组数据进行分析,分别获取了21835502、21762356和22037888个Clean Reads,GC含量分别为43.75%、43.96%和43.99%。且3个不同器官的Q30碱基百分比均在93.19%以上,说明转录组测序质量较高,满足后续试验要求(表1)。
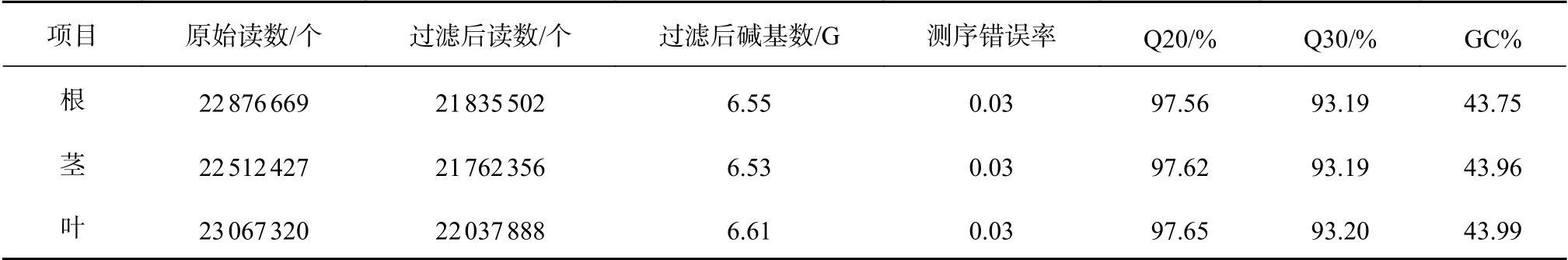
表1 样本测序数据质量汇总Table 1 Quality summary of sample sequencing data
2.1.2 差异基因数目统计
利用Edge R软件,以表达量差异倍数大于1和Padj≤ 0.005为差异基因筛选标准,对国槐根、茎、叶3个不同器官的转录组数据进行比对(图1)。结果表明,根与叶中差异基因数最多,为52711条;根与茎次之,为51937条;茎与叶最少,为7193条。
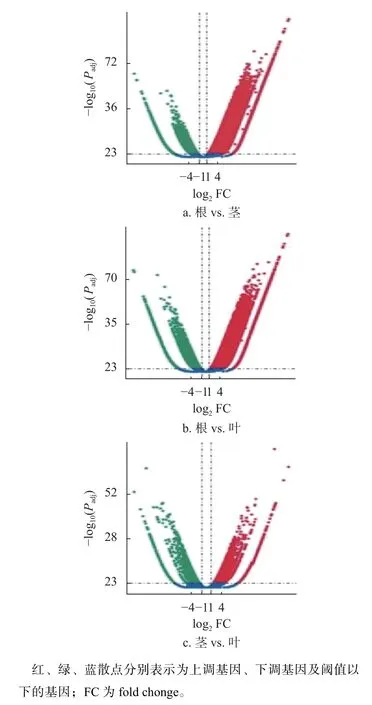
图1 国槐不同器官差异表达基因火山图Fig. 1 Differential expression of genes in different organs of S. japonica
2.1.3 差异基因GO功能富集
GO (Gene Ontology) 数据库包括生物过程、细胞组成和分子功能三大生物学过程。本试验采用Cluster Profiler软件对差异基因进行GO功能富集分析,阈值为Padj< 0.05。
图2分别为各比较组中差异基因的GO富集结果,在根vs.茎比较组中(图2a),注释到GO数据库的差异基因数目为28755条,分属于3009个生物学过程、684个细胞组分和1456个分子功能组分,主要显著富集于代谢过程、蛋白质代谢过程及高分子化合物等功能;在根vs. 叶比较组中(图2b),注释到GO数据库的差异基因数目为29352条。分属于3024个生物学过程、692个细胞组分和1462个分子功能组分中,主要显著富集于蛋白质代谢过程、膜的外在成分及四吡咯结合等功能;在茎vs. 叶比较组中(图2c),注释到GO数据库的差异基因数目为4440条,分属于2269个生物学过程、536个细胞组分和1019个分子功能组分中,主要显著富集于代谢过程、膜部分及催化活性等功能。
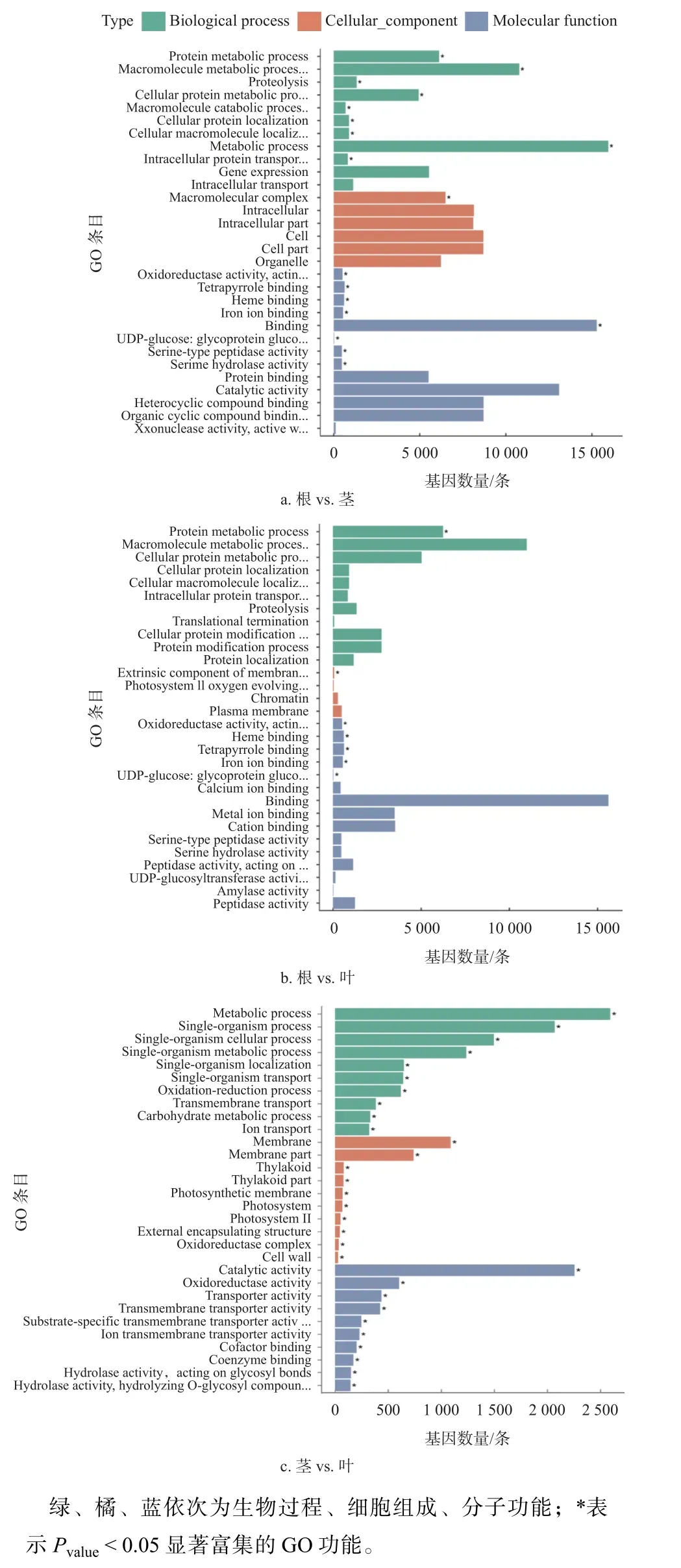
图2 GO 分类的显著性富集图Fig. 2 Significant enrichment plots of GO terms
2.1.4 差异基因KEGG功能分析
KEGG数 据 库(Kyoto Encyclopedia of Genes and Genomes) 是整合了基因组、化学和系统功能信息的综合性数据库,本研究以Padj< 0.05为阈值,进行差异代谢通路富集分析。在根vs. 茎(图3a)及根vs. 叶(图3d)比较组中,分别有13961、14159条差异基因被注释到了123个代谢通路,但均未被显著富集。在茎vs. 叶(图3g)比较组中,有2056条差异基因被分类到117个代谢通路中,其中糖酵解和糖质新生、苯丙烷类生物合成及乙醛酸和二羧酸代谢的富集达到了显著水平(P<0.01)。
进一步分别对3个比较组中的上调及下调基因进行KEGG富集,在根vs. 茎(图3b,c)及根vs.叶(图3e,f)比较组中,其上调基因在核糖体代谢通路中的富集达到了显著水平(P< 0.01),其下调基因在光合作用,光合作用−天线蛋白等通路中的富集达到了显著水平(P< 0.01);在茎vs. 叶(图3h,i)比较组中,其上调基因在苯丙烷生物合成、糖酵解/糖异生、戊糖磷酸途径等代谢通路中的富集达到了显著水平(P< 0.01),下调基因在光合作用、光合作用−天线蛋白、光合生物的固碳作用等通路中的富集达到了显著水平(P< 0.01)。

图3 KEGG pathways富集图Fig. 3 Enrichment plots of KEGG pathways
2.2 国槐EST-SSR引物开发
2.2.1 不同组织转录组SSR位点的数量及分布
分别对国槐根、茎、叶的转录组数据进行组装(表2)。结果表明,根的Unigene数最多,茎组织次之,叶组织最少。各组织转录组SSR位点检测结果表明,茎组织中SSR位点数最多,叶组织次之,根组织最少。本研究中国槐的SSR位点类型丰富,单核苷酸至六核苷酸重复类型均存在,其中单核苷酸重复占主导地位,说明国槐转录组中的SSR重复单元主要为单碱基重复。
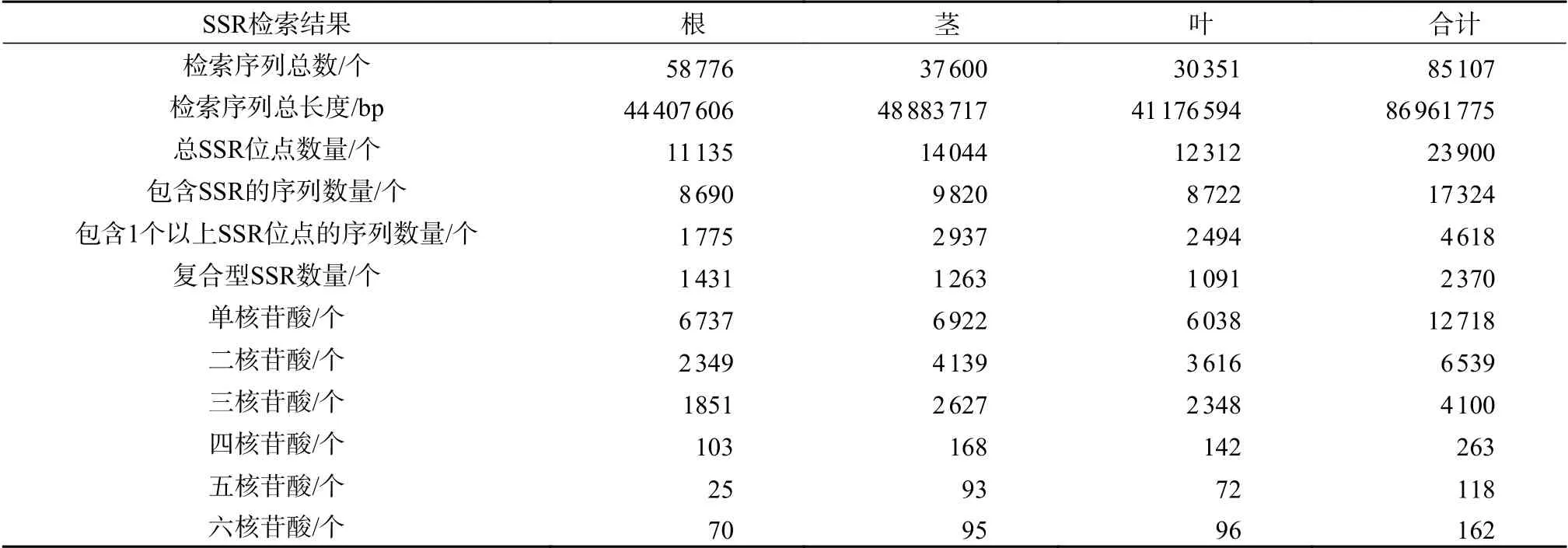
表2 国槐SSR检索结果Table 2 SSR search results of S. japonica
2.2.2 EST−SSR重复类型及频率特征
分别对国槐根、茎、叶不同器官及全部样本进行SSR序列长度分析(图4),结果表明,国槐不同组织及全部样本的SSR序列长度在10~81 bp间变化,主要以10~22 bp短序列为主。分别占不同组织及全部样本SSR总数的89.00%、90.06%、89.70%及90.17%,其中10 bp的SSR数量最多,分别占各自SSR总数的20.80%、18.05%、17.83%及20.05%,且主要以单核苷酸重复为主。
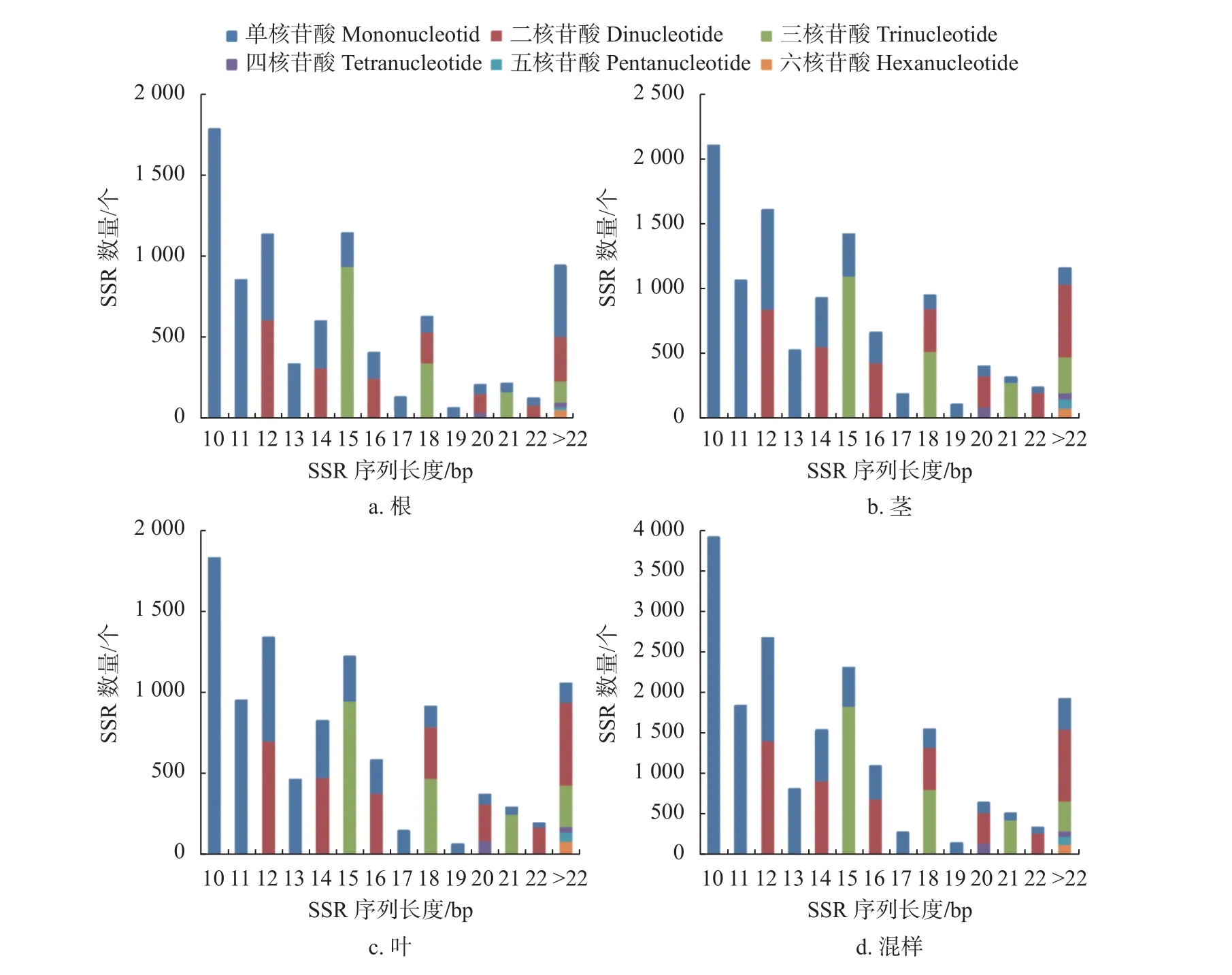
图4 SSR序列长度分布Fig. 4 SSR sequence length distribution
进一步对各器官中的重复单元及优势重复模序进行统计分析(表3~4),结果表明,国槐根、茎、叶及全部样本SSR位点分别含有217、300、286、403种重复单元。在根、茎、叶不同器官的转录组中,茎检测到的重复模序种类最多,根最少。对各样本SSR重复模序的类型进行统计,结果表明,各样本中单、二、三核苷酸重复的优势模序均为A/T、 AT/TA和AAG/CTT,四、五、六核苷酸重复模序虽种类较多,但数量相对较少。
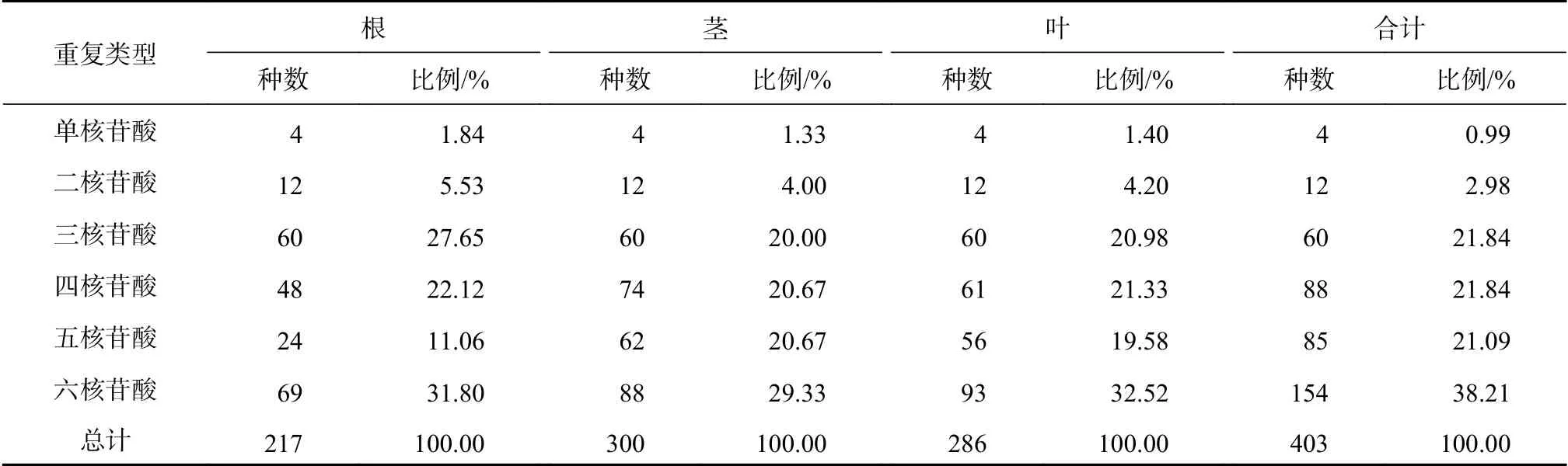
表3 国槐SSR重复类型统计Table 3 SSR repeat type statistic of S. japonica

表4 SSR优势重复模序比例Table 4 Proportion of preponderant repeat motif of SSR
2.2.3 国槐EST−SSR引物有效性及多态性
依照国槐3个不同器官的转录组混合拼接数据,依据筛选标准设计引物,随机挑选出符合条件的30对,以11个国槐无性系的DNA为模板,进行有效性验证。共筛选出9对多态性引物(表5~6),多态性带数百分比均值为 79.63%,各引物的多态信息指数(PIC)值在0.1528~0.6719之间变化,Shannon's 信息指数(I)在0.1849~1.1384之间变化,变幅均较大,说明筛选出的引物多态性较高,但不同SSR位点的遗传信息存在较大差异,部分引物的扩增结果见图5、图6。
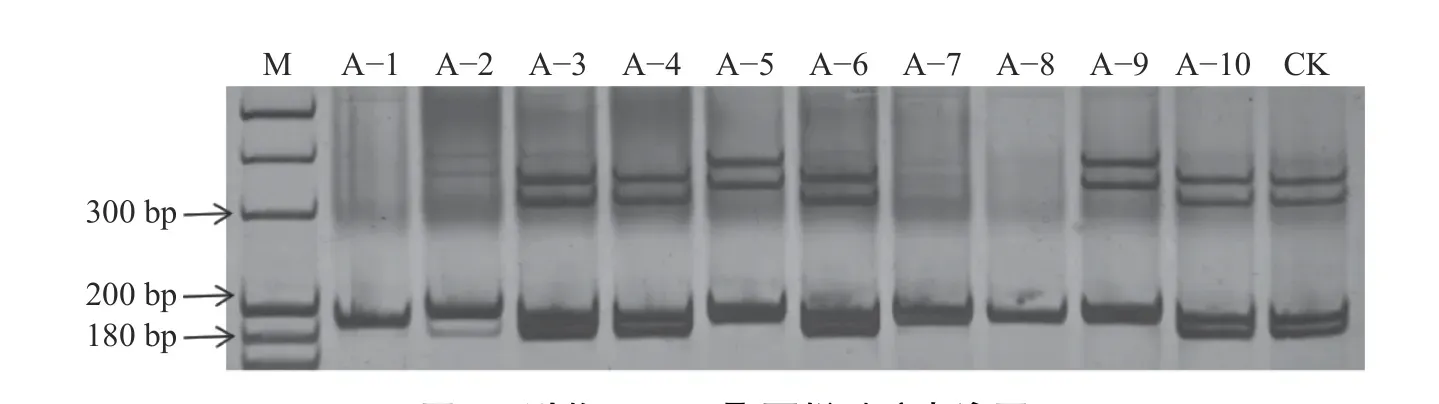
图5 引物YJ-23聚丙烯酰胺电泳图Fig. 5 Primer YJ−23 polyacrylamide electrophoretogram
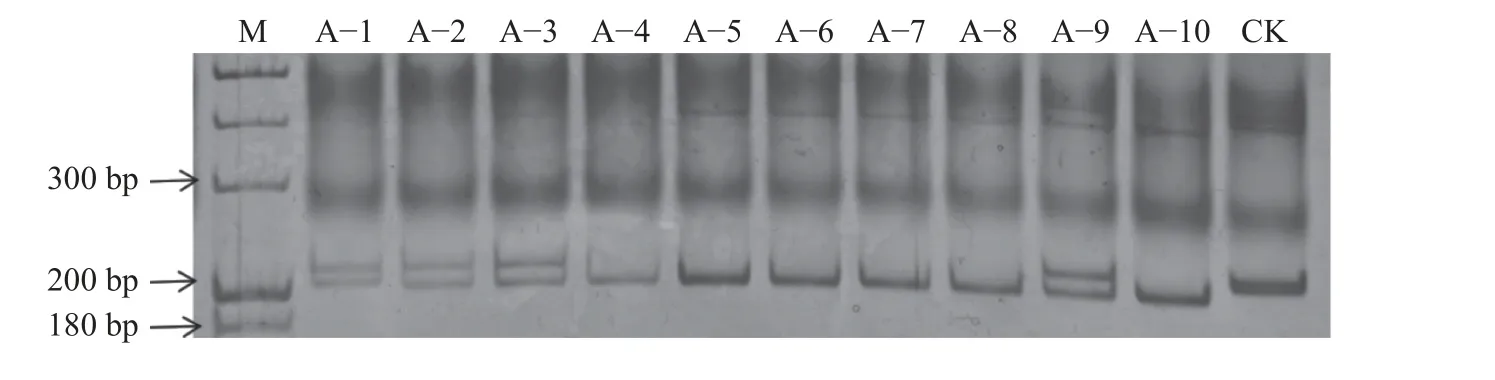
图6 引物YJ-27聚丙烯酰胺电泳图Fig. 6 Primer YJ−27 polyacrylamide electrophoretogram

表5 9对多态引物基本信息Table 5 Information of 9 pairs of polymorphic primers
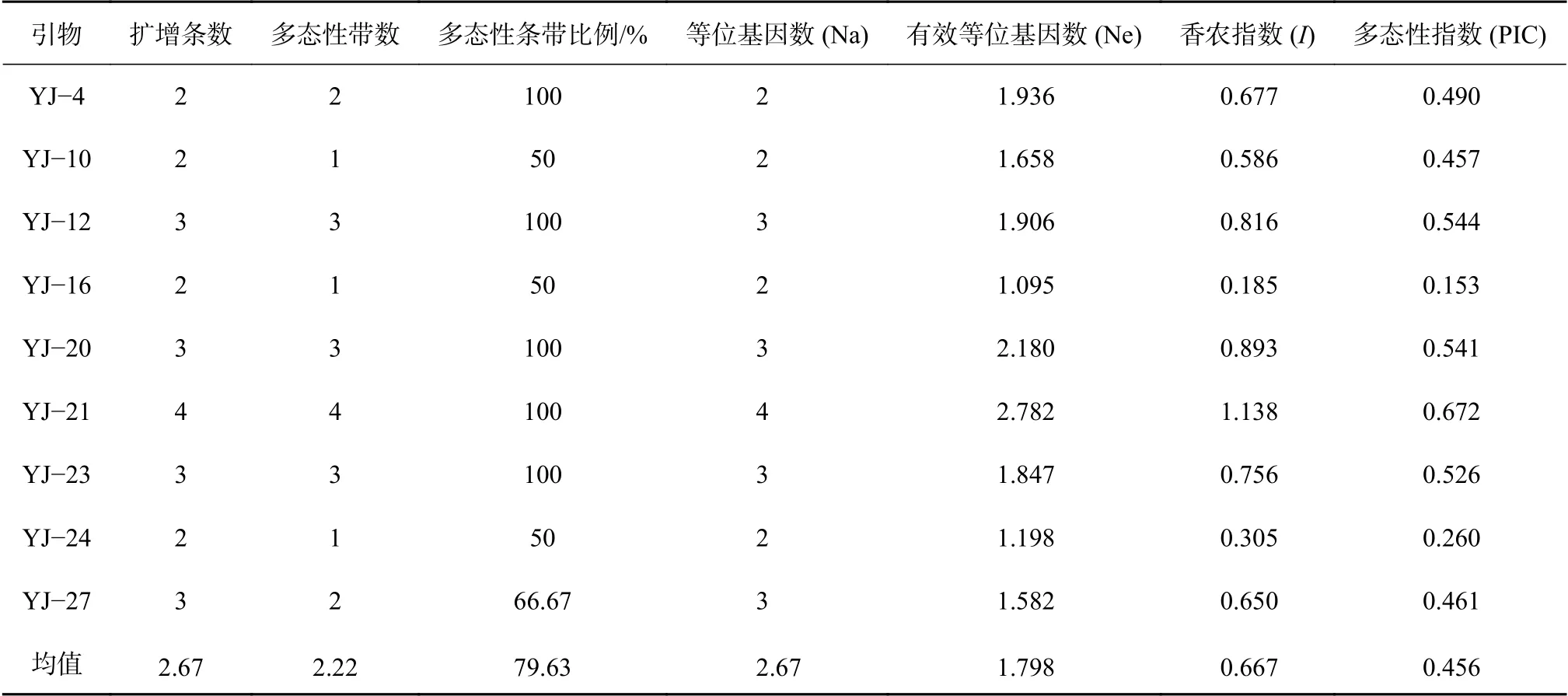
表6 9对引物扩增结果Table 6 Amplification results of 9 pairs of primers
2.2.4 不同国槐无性系的进化树及指纹图谱
依据各引物的扩增结果,将条带转化为二元数据(0/1 矩阵),运用DPS 7.05 数据处理系统(UPGMA 类平均法),计算各样本间的遗传距离矩阵并绘制11个不同国槐参试无性系进的进化树,构建指纹图谱(图7、表7),结果表明,本研究开发的9对EST−SSR引物可将全部参试样本完全区分,当遗传距离为0.415时,可按遗传关系的远近将全部参试样本分为4类,第Ⅰ类包括CK、A−4、A−6、A−8、A−1 第Ⅱ类包括A−3、A−9、A−5、A−7;第Ⅲ类包括 A−2 ;第Ⅳ类为 A−10,这也进一步验证了引物的可靠性。
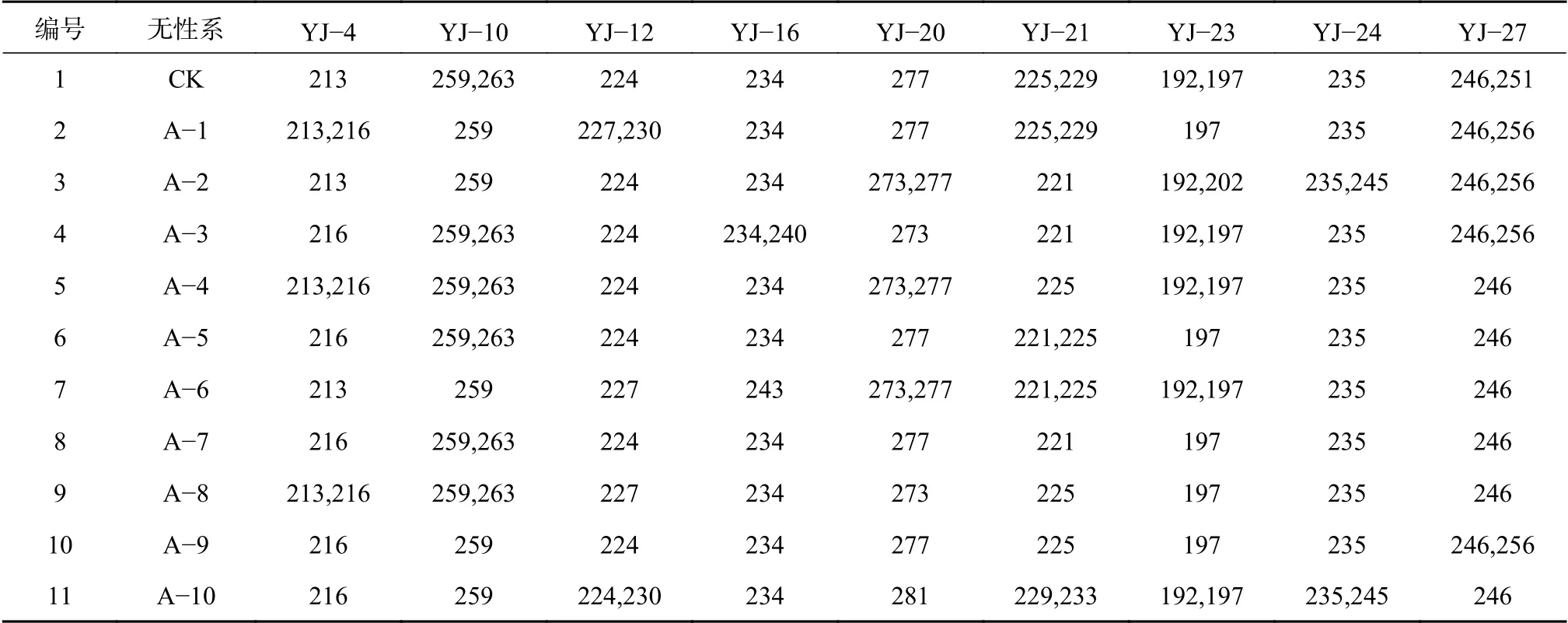
表7 11个国槐无性系的EST-SSR指纹图谱Table 7 SSR fingerprint of 11 S. japonica clones
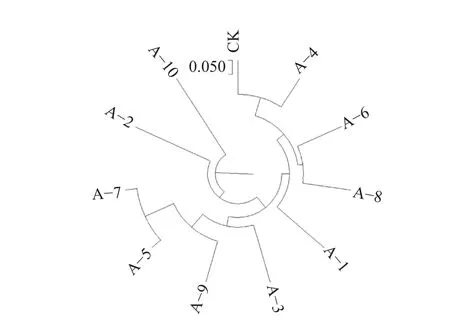
图7 国槐11个不同无性系SSR聚类分析Fig. 7 SSR cluster analysis diagram of 11 different clones of S. japonica
3 结论与讨论
Illumina高通量测序技术,作为第二代测序技术,兼具高通量、效率高及成本低等诸多优点,近年来已在生命科学领域得到广泛应用[19]。本研究利用 Illumina 高通量测序技术对国槐根、茎及叶3个不同器官进行了无参转录组测序,转录组对比结果表明,根vs. 茎比较组中的差异基因数最多,根vs. 叶次之,茎vs. 叶最少,这与王芬[20]等得出的福鼎大白种茶(Camellia sinensis)树根与叶相比差异基因数最多的结论不同,原因可能是由于树种或筛选阈值设定不同导致的。进一步对差异基因进行GO和KEGG注释,结果表明,根vs. 茎、根vs. 叶、茎vs. 叶3个比较组中差异基因的GO及KEGG数据库注释率均值分别为57.59%和27.44%,二者虽均有大量差异基因未获得注释,但GO数据库的注释率远高于KEGG数据库,这与刘洪旭等[21]、葛国平等[22]众多学者的研究结果相似,原因可能与物种及数据库的侧重点不同有关,同时测序时参考基因组的有无也可能对后续分析的产生影响[23]。
本研究表明,在国槐根、茎、叶器官的转录组数据两两对比中,每个比较组的差异基因GO富集情况大体一致。在生物学过程中,主要参与代谢过程,说明国槐不同器官均存在大量基因参与次生代谢过程;在细胞组分过程中,主要构成细胞的组成;在分子功能过程中,多数具有结合功能和催化活性等功能,说明植物生长后期酶活催化能力增强。这与林艳玲等[24]在对人参(Panax ginseng)根、茎、叶转录组两两对比,得出的差异基因GO富集结果相似,表明不同植物在生长发育过程中均受到多种途径及多种代谢产物的影响。同时在茎vs. 叶比较组中,有10个被KEGG数据库显著富集的代谢通路,但在根vs茎和根vs叶比较组中,均未有代谢通路被显著富集,原因可能是由于植物DNA在不同组织中的转录过程存在差异,从而导致转录产物的不同或表达量存在差异[25],也可能是由于植物部分组织中的DNA存在甲基化现象,从而抑制了某些功能基因的表达[26],但有待进一步验证。进一步对各比较组中上下调基因进行KEGG富集分析,结果表明,从宏观上来看,上调基因主要在核糖体代谢通路中被显著富集,下调基因主要在光合作用、光合作用−天线蛋白等代谢通路中被显著富集,原因可能是由于上调基因主要参与细胞分裂及组织分化等过程[27],而由于根位于地下部分,茎叶位于地上部分,与光合作用相关的基因不升反降,原因可能是由于这些基因的表达量负相关于基因功能[28]。同时,结果表明,在根vs. 茎、根vs. 叶2个比较组中上调基因数量远多于下调基因,原因可能是由于此时植物的光形态建成已趋于完成,且叶绿素含量已趋于稳定,而此时期为国槐速生期,与植物生长相关的基因转录、表达较为活跃,进而使得细胞分裂加快,同时也提高了植物组织分化的速度[29−31]。各组织转录组的SSR分析表明,茎组织的SSR位点数最多,叶组织次之,根组织最少,这与石登红等[32]在对野地瓜(Ficus tikoua)不同组织转录组的研究中得出的茎SSR位点数多于叶片的结论一致;对各样本SSR重复模序的类型进行统计,结果表明,各样本三核苷酸重复的优势模序均为AAG/CTT,这与众多国内外学者[33−35]对双子叶植物SSR模序分析所得结论一致,也进一步说明了AAG/CTT 微卫星基序在双子叶植物中的广谱性及重要性。进一步利用国槐3个不同器官的转录组混拼数据,按照严格的标准,进行EST-SSR引物的开发,结果表明,在随机挑选的30对引物中,有24对引物能扩增出清晰的目的条带,进一步的验证表明,其中有9对多态性引物,多态性条带比例高于Sun等[14]开发的国槐SRAP引物,但SRAP标记也具有其自身优势,与SSR分子标记相比其扩增范围包括了DNA的内含子区域,扩增范围更加广泛,但SSR分子标记具有更高的种间通用性及精确性[36],未来将2种技术结合进行遗传多样性分析,不失为解决引物资源缺乏、提高分子育种效率的良策。
本研究对国槐的根、茎、叶进行了转录组测序,对3个不同器官的转录组数据进行两两对比,对差异基因进行了功能注释及代谢通路分析,同时对不同样本的EST−SSR进行了分析,开发出了9对多态性高的EST−SSR引物,同时构建了11个不同国槐无性系的进化树及指纹图谱,为今后国槐的遗传资源开发及品种鉴定等工作提供了重要的分子依据,同时为今后类似的研究提供了参考。
猜你喜欢
动物医学进展(2024年4期)2024-04-10 01:50:04
现代园艺(2021年8期)2021-12-06 14:52:08
心电与循环(2020年1期)2020-02-27 07:48:24
诗潮(2017年12期)2018-01-08 09:46:04
延河·绿色文学(2016年8期)2016-05-14 06:40:42
山西林业(2015年4期)2015-12-25 16:09:55
湖北农业科学(2014年3期)2014-07-21 10:25:00
云南中医学院学报(2011年4期)2011-07-31 17:54:43
中学生数理化·八年级数学华师大版(2008年3期)2008-08-26 11:26:16
现代农业科技(2006年23期)2007-01-05 03:23:54
