
α-Fe2O3催化臭氧氧化处理苯酚废水的效果及机理
2022-08-25 13:59杜明辉高群丽张耀宗孙晓明
环境科学研究 2022年8期
王 勇,杜明辉,张 宁,高群丽,张耀宗*,孙晓明*
1. 华北理工大学建筑工程学院,河北 唐山 063000
2. 中国环境科学研究院,国家环境保护生态工业重点实验室,北京 100012
苯酚是石化、制药和印染等行业重要的生产原料、中间产物或废弃物[1],是具有代表性的高毒性、难降解污染物,常规生物法难以将其去除,已对我国污水厂的废水处理造成沉重负担[2]. 催化臭氧氧化工艺已被广泛应用于污水厂末端的深度处理[3-5],用以去除废水中的难降解污染物,该工艺主要利用在催化过程产生的羟基自由基(·OH)[6],对有机物进行无选择性矿化,特别是对苯酚等毒性有机物表现出稳定的去除效果[7]. 然而,催化臭氧氧化工艺仍存在诸如催化效率较低、催化剂易中毒失活、活性组分涂覆不均等问题亟待解决[8-10]. 因此,选取适应性广泛、持续催化能力强、活性组分稳定的催化剂极为重要. 有研究表明,粉态金属氧化物如铁基、锰基等催化剂因其具有结构稳定、易于合成等优点逐渐受到关注[11-14],此类催化剂可以近似均相地参与臭氧催化,能够为臭氧和有机物的吸附分解提供充足的反应点位[15-16],提高·OH的产率,较常规负载型催化剂显著增强催化效率. Wu等[17]通过吡啶吸附/解吸的红外光谱研究,使金属氧化物上的Lewis和Bronsted酸位点得以定量,结果表明,几种材料的Lewis酸位点总数排序依次为α-Fe2O3> CuO > γ-Al2O3> ZnO > CaO. Magario等[18]使用铁基催化剂重点讨论了催化臭氧氧化过程的自由基生成机理,发现α-Fe2O3较同类型材料可以为催化臭氧氧化提供更加充足的反应位点. 由于上述特性,α-Fe2O3存在高比表面积和暴露的表面活性位点,具备催化臭氧的应用潜力. He等[19]在类芬顿条件下去除难降解有机物,进一步证实了α-Fe2O3具有较强的臭氧分解活性和稳定性[20-21],这表明α-Fe2O3是可被应用于催化臭氧氧化的优良催化剂,但α-Fe2O3对难降解废水的催化臭氧氧化效果和反应机制有待充分研究.
该研究以苯酚作为模拟污染物,通过对比试验及单一变量试验,考察α-Fe2O3催化臭氧氧化苯酚的去除效果,探究了反应的影响因素和反应动力学,以期为寻找难降解有机废水高效且经济的处理工艺提供技术参考;同时,通过·OH屏蔽及反应位点试验,深入讨论了·OH的生成机制和反应活性位点,测定了α-Fe2O3在循环使用过程中的稳定性,以期为实际应用中建立完善的难降解废水处理工艺体系提供一定的经验.
1 材料与方法
1.1 试验材料
试验用催化剂α-Fe2O3、α-MnO2、TiO2、MgO、γ-Al2O3的粒径均约为6 μm,使用去离子水配制50 mg/L苯酚溶液. 试验试剂主要包括苯酚(分析纯)、纯氧(99.999%)、盐酸(分析纯)、氢氧化钠(分析纯)、叔丁醇(分析纯)、去离子水等.
1.2 试验装置
催化臭氧氧化工艺流程如图1所示. 反应器直径70 mm,高600 mm,有效容积2 L. 通过臭氧浓度检测器(3S-J5000,北京同林科技有限公司)检测气相臭氧浓度,试验开始前先开机预热,控制仪器温度20~24 ℃. 通过臭氧发生器(3S-T,北京同林科技有限公司)用纯氧制备臭氧,调节臭氧发生器功率控制臭氧浓度,尾气通过硫代硫酸钠溶液吸收. 装置底部放置转子进行磁力搅拌,防止催化剂沉积. 除特别说明外,试验采用参数均为流量100 mL/min、臭氧投加量10 mg/L、催化剂投加量0.5 g/L、初始pH7,反应时间1 h,每5 min取样一次. 检测水样COD、pH、溶解态臭氧浓度及紫外光谱,分析α-Fe2O3催化臭氧氧化废水效果.
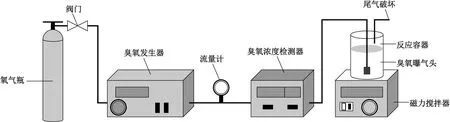
图1 催化臭氧氧化工艺流程Fig.1 Catalytic ozone oxidation process flow chart
1.3 表征及检测方法
用X射线衍射仪(XRD,布鲁克,D8 advance,德国)对α-Fe2O3物相定性,扫描角度2θ为10°~70°,扫描速度为8 °/min. 通过扫描电子显微镜(SEM,蔡司evo18,德国)得到α-Fe2O3的表面形态和结构. 120 ℃下干燥5 h预处理α-Fe2O3后,使用全自动比表面及孔隙度分析仪(BET,麦克,ASAP2020,美国)在氮气气氛下测定α-Fe2O3比表面积及内部孔径,采用BET(Brunauer-Emmett-Telle)方程计算比表面积,采用BJH(Barrett-Joyner-Hallender)吸附模型计算α-Fe2O3孔径大小及分布.
采用多参数水质测定仪(LH-3BA,北京连华永兴科技发展有限公司)测定废水COD浓度;采用紫外分光光度计(UV-8000S,上海元析仪器有限公司)检测波长为200~400 nm的废水紫外光谱,考察污染物结构的变化情况;采用DPD标准方法(SPH006CN+,百灵达Palintest,英国)检测液相臭氧浓度;采用pH计(PHS-2F)测定溶液pH;使用全普直读型电感耦合等离子光谱仪(ICPE-9820,Shimadzu,日本)检测溶液中Fe2+和Fe3+的浓度.
2 结果与讨论
2.1 α-Fe2O3表征
α-Fe2O3具有较好的臭氧催化性能,对其进行XRD、SEM、N2吸附脱附表征,结果如图2~4所示.图2为α-Fe2O3的X射线衍射图谱,在2θ为24.14°、33.12°、35.60°、40.82°、49.42°、54.04°、57.56°、62.38°、63.96°处均出现衍射峰值,通过MDI Jade 6软件进行标准PDF卡片查找,检索后Fe2O3显示为最小FOM值(2.3),衍射峰对应(012)(104)(110)(113)(024)(116)(018)(214)(300)晶面. 图3为α-Fe2O3的SEM扫描电镜形貌. 由图3可见,α-Fe2O3呈团聚的不规则球状.
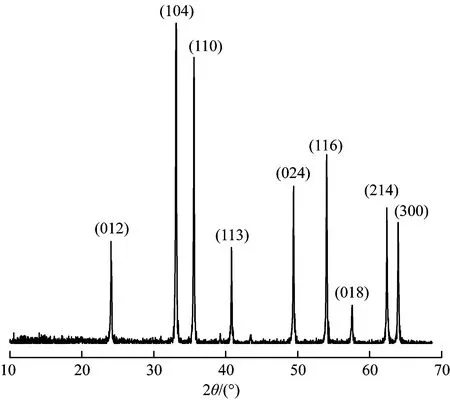
图2 α-Fe2O3的X射线衍射图谱Fig.2 X-ray diffraction pattern of α-Fe2O3

图3 α-Fe2O3的扫描电镜形貌Fig.3 Scanning electron microscopic morphology of α-Fe2O3
α-Fe2O3的N2吸附/脱氮等温线如图4(a)所示,曲线符合Ⅳ型等温曲线特征. 在较高相对压强(P/P0)区,由于毛细作用凝聚产生了H1型滞留环;在低P/P0区,α-Fe2O3的N2吸附为单层吸附且吸附量较少,这说明α-Fe2O3存在介孔结构. 图4(b)为α-Fe2O3的孔径分布曲线. 由4(b)可见,α-Fe2O3的孔径主要分布在1.82~10.2 nm之间. 采用BET方程及BJH吸附模型计算α-Fe2O3比表面积、孔体积及平均孔径,其值分别为83.38 m2/g、0.135 cm3/g、7.447 nm.
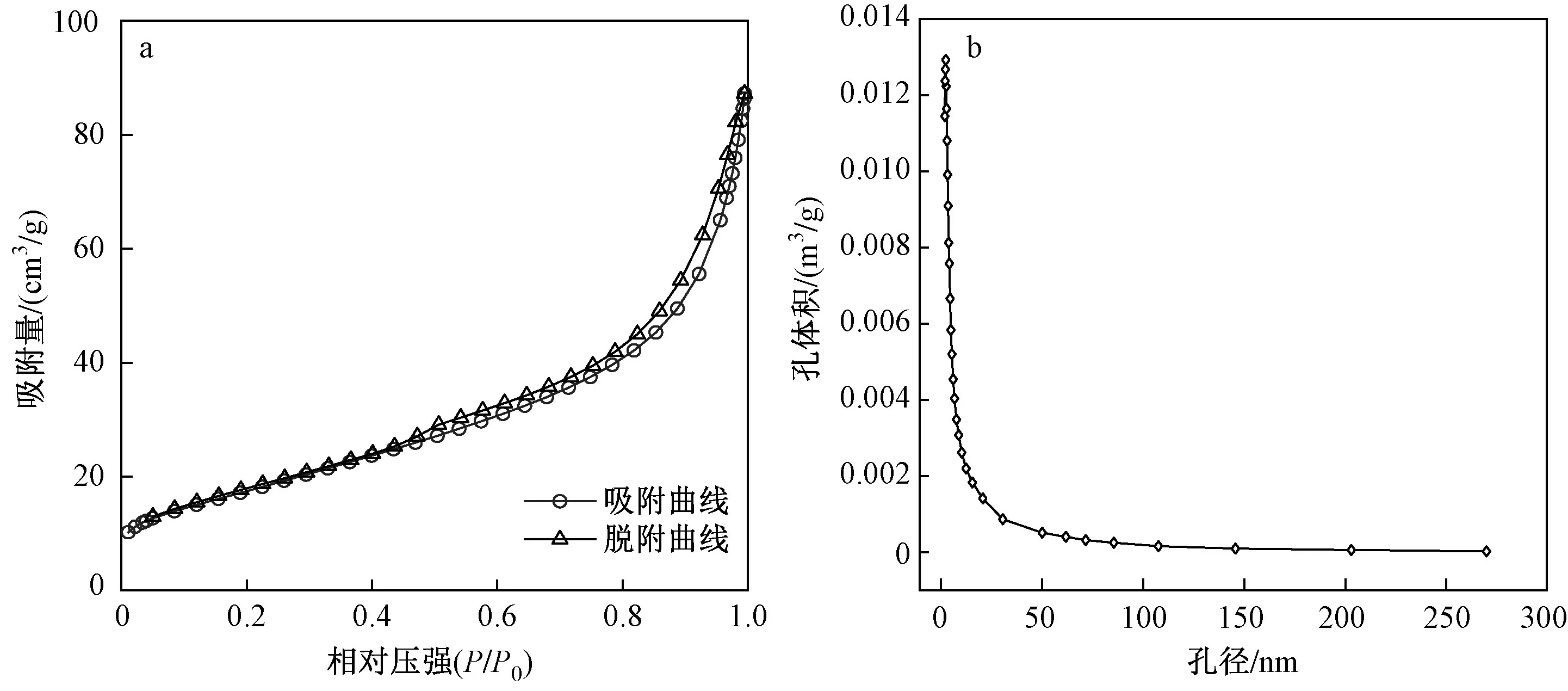
图4 α-Fe2O3的N2-吸附/脱附曲线及孔径分布Fig.4 N2-adsorption/desorption curve and pore size distribution of α-Fe2O3
2.2 催化剂的臭氧催化性能
研究表明,α-Fe2O3[24]、MgO[25]、[22]、α-MnO2[23]、TiO2γ-Al2O3[26]均具有较高的臭氧催化活性. 为研究α-Fe2O3与同类型催化剂的臭氧催化性能差异,对催化臭氧氧化体系下不同催化剂对苯酚废水COD的降解效果(见图5)和反应动力学(见表1)进行了对比和分析. 由图5和表1可见,伪一级动力学拟合参数R2均大于0.980,这说明拟合结果较好. 反应结束时各催化剂的降解效果表现为α-Fe2O3(97.67%)>α-MnO2(87.21%) > MgO (85.66%) > TiO2(84.37%) >γ-Al2O3(78.18%)>对照组(69.11%). 对于单独臭氧氧化体系,臭氧对苯酚废水COD的去除在反应40 min后趋于平缓,反应速率仅有0.024 s−1,因此单独臭氧氧化体系对苯酚废水COD的去除能力有限. 而对于催化臭氧氧化体系,苯酚废水COD的去除效果存在不同程度的提升,值得注意的是,从COD的去除率和反应速率常数均可以看出,投加α-Fe2O3催化臭氧氧化效果最好,反应30 min时已达到最佳的COD去除效果,且反应的速率常数明显高于其他对照试验,较其他催化过程显著提高了污染物的矿化水平. 由此可见,α-Fe2O3较其他催化剂有较大优势,这可能是由于α-Fe2O3对臭氧具有更强的催化活性和对反应环境更好的适应性[17,27].

图5 催化臭氧氧化体系下不同催化剂对苯酚废水COD的降解效果Fig.5 Effect of different catalysts on COD degradation of phenol wastewater under catalytic ozonation system

表1 伪一级动力学的拟合参数Table 1 Fitting parameters of pseudo first order dynamics
为进一步检验苯酚废水在α-Fe2O3催化臭氧氧化和单独臭氧氧化过程的降解情况,需要对反应过程的水样进行紫外-可见光光谱扫描[28]. 苯酚在紫外光270 nm附近表现为特征吸收峰,且在降解过程中于270 nm附近形成吸收带. 因此,选取200~400 nm作为扫描范围. 如图6所示,随着反应进行,催化臭氧氧化和单独臭氧氧化两种体系下苯酚的特征吸收峰均逐渐降低,说明苯酚被逐渐氧化. 反应10 min后,在200~300 nm处有明显吸收带,表明体系中苯环及中间产物的存在,说明苯酚发生了邻位开环反应[29],生成了邻苯二酚、对苯二酚、间苯二酚及苯醌等中间产物. 反应30 min时,单独臭氧氧化体系在270 nm处特征峰消失,但仍具有一定的吸收带,而催化臭氧氧化体系的吸收带均消失,这说明苯酚废水几乎被完全降解.
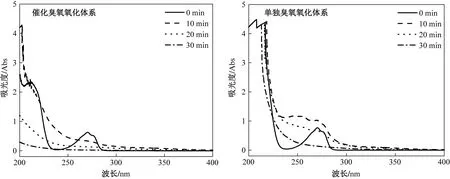
图6 α-Fe2O3催化臭氧氧化和单独臭氧氧化体系下苯酚废水的紫外光谱Fig.6 UV spectra of phenol wastewater under α-Fe2O3 catalytic ozonation and ozonation alone system
除了臭氧的直接氧化作用和催化臭氧而产生·OH的氧化作用外,苯酚废水COD的去除效果还可能与α-Fe2O3的吸附性有关,α-Fe2O3在不同投加量下对苯酚废水COD的吸附过程如图7所示. 由图7可见,随着α-Fe2O3投加量的增加,各反应分别在25、15、10 min达到吸附平衡,但苯酚废水COD的吸附效果没有明显提升. 在α-Fe2O3投加量为0.1 g/L时,苯酚废水COD的去除率为7.03%,而α-Fe2O3投加量提高至5 g/L时,COD的去除率仅提高了4.79%. 已有研究[30]表明,α-Fe2O3的吸附性有利于臭氧与α-Fe2O3接触分解以及污染物的快速去除,而α-Fe2O3吸附对苯酚废水COD去除的影响微弱,在较低α-Fe2O3投加量时吸附影响可忽略不计. 这说明α-Fe2O3的催化臭氧氧化过程并不依赖吸附对反应的控制,有可能是由于α-Fe2O3表面具有较多的活性位点,以在低吸附能力下催化臭氧产生·OH,进而高效去除污染物.
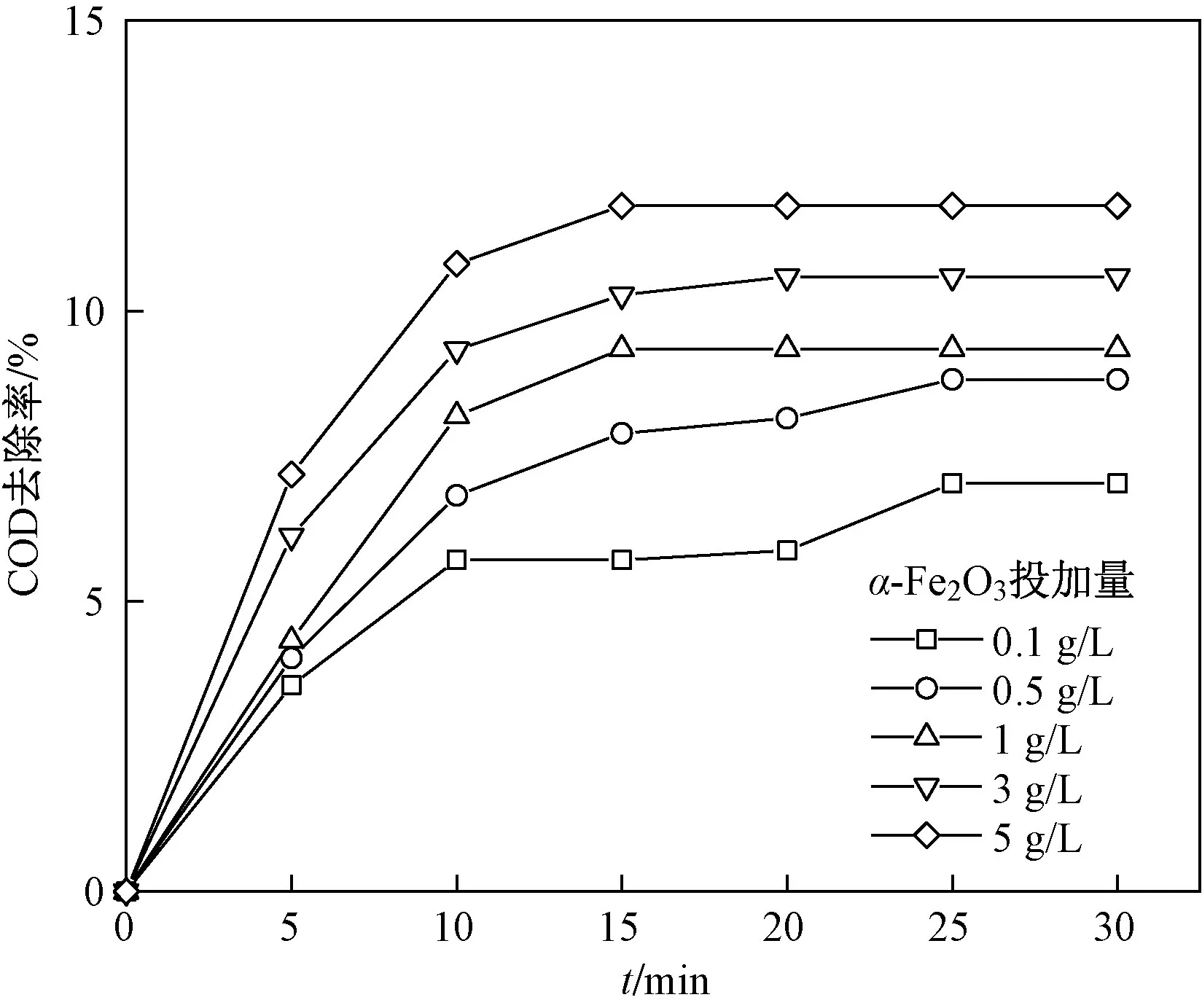
图7 不同α-Fe2O3投加量对苯酚废水COD的吸附性比较Fig.7 Adsorption comparison of COD degradation phenol with different α-Fe2O3 dosages
2.3 反应条件优化
有研究[31]表明,臭氧投加量、催化剂投加量、pH是催化臭氧氧化过程的主要影响因素. 参数选取对催化臭氧氧化过程和污染物氧化效果影响明显. 为优化α-Fe2O3催化臭氧氧化运行工况,通过单一变量试验考察上述因素对催化臭氧氧化过程的控制情况,选取氧化效果更好且经济的反应条件.
2.3.1 臭氧投加量
臭氧是反应过程主要的氧化剂和氧化剂前体,保证一定水平的臭氧投加利于反应的快速进行,但过量的臭氧会造成资源浪费,有必要对臭氧用量进行优化. 如图8所示,在α-Fe2O3投加量为0.5 g/L、pH为7的条件下,COD的去除率随着臭氧投加量增加而明显提高. 臭氧投加量为2.25 mg/L时,试验结束后COD的去除率仅为58.13%;臭氧投加量为5 mg/L时,反应60 min时COD的去除率最大,为97.62%;臭氧投加量为10 mg/L时,仅需30 min即达到相同水平的去除效果. 这说明初步增加的臭氧大大提高了其对α-Fe2O3和污染物的接触机会,明显加快了降解效率. 但进一步提高臭氧投加量至15 mg/L时,其对污染物的降解效果提升不明显,这是因为溶液存在一定饱和平衡浓度,增加的臭氧并未被有效利用. 因此,将10 mg/L作为后续试验的臭氧投加量.

图8 不同臭氧投加量对COD去除率的影响Fig.8 Effect of different ozone dosage on COD removal rate
2.3.2 催化剂投加量
催化剂活性位点的数量影响污染物的去除效果[32]. 一般来说,随着催化剂用量的增加,反应位点和反应表面积增加;同时,将获得更高的催化臭氧化表观反应速率常数. 然而,考虑到初始污染物浓度恒定,过量的催化剂会降低单位面积污染物和臭氧浓度,从而降低催化效率. 为探究α-Fe2O3投加量对催化臭氧氧化过程的影响,选取了α-Fe2O3投加量梯度进行了催化效果试验,试验设置臭氧投加量为10 mg/L,pH为7. 如图9所示,改变α-Fe2O3投加量对苯酚废水COD最终的去除效果差别不大. α-Fe2O3投加量为0.5 g/L时表现最佳,反应30 min苯酚几乎完全矿化.相同时间下,α-Fe2O3投加量为0.01、0.05、0.1、1 g/L时,COD去除率也达到较高水平. α-Fe2O3投加量为1 g/L时,COD的去除率略有下降,此时的α-Fe2O3可能已经过量,这与Wang等[25]的结果相似.因此,粉态α-Fe2O3体现了明显的催化活性,在低α-Fe2O3投加量下也能够发挥臭氧催化作用,而过量的α-Fe2O3不利于反应进行.

图9 不同α-Fe2O3投加量对COD去除率的影响Fig.9 Effect of different α-Fe2O3 dosage on COD removal rate
2.3.3 初始pH
介质pH控制着α-Fe2O3表面电性和反应介质酸碱性[33],进而影响臭氧的分解,α-Fe2O3表面电荷高于或低于零点电位(pHpzc)时,·OH的产生和吸附过程也会受到影响[34]. 研究了不同初始pH条件下COD的去除情况,设置臭氧投加量为10 mg/L,α-Fe2O3投加量为0.5 g/L,试验结果如图10所示. 由图10可见,COD的去除率随着反应时间的延长逐渐升高,反应60 min后均达到最大值,但中性条件较酸性和碱性条件表现了更高的苯酚废水COD降解效率. pH=7时废水处理效果最佳,反应30 min时COD的去除率已稳定达到97.51%. 在pH=3时,氧化过程受到明显抑制,反应结束后去除率仅为63.39%. 这是因为酸性条件下臭氧分子较为稳定,氧化过程以臭氧的环加成反应和亲电取代反应为主,这在一定程度上延缓了臭氧转化为·OH. 另外,碱性条件有利于臭氧的分解,促进·OH的生成,但pH为9和12时,反应速率较中性条件反应速率下降,COD的去除率分别降低了4.72%和15.12%. 这可能是由于催化剂表面电性偏离了pHpzc,亲电基团减少,从而削弱了催化剂与臭氧的相互作用,导致·OH生成速率降低. 因此,中性条件更接近α-Fe2O3的pHpzc,在反应过程中保持中性的反应介质可以保证最佳的污染物降解效率,最能发挥催化剂的催化效果.

图10 不同初始pH对COD去除率的影响Fig.10 Effect of different initial pH on COD removal rate
2.4 α-Fe2O3对臭氧传质的影响
催化剂可以加速臭氧的传质作用,加快臭氧分解,这一过程促进了·OH的产生. 为研究α-Fe2O3对臭氧的分解促进作用,进行了臭氧传质试验. 图11(a)为溶解性臭氧在反应过程中随时间的变化曲线,两条曲线表示自然状态和加入α-Fe2O3后臭氧的分解情况.单独臭氧在20 min时达到臭氧平衡浓度(12.7 mg/L),而投加α-Fe2O3时较单独臭氧的臭氧平衡浓度降低了33.86%,且提前5 min达到浓度平衡. 停止通入臭氧后,臭氧的分解速率可以由伪一级动力学表达.图11(b)为投加和未投加α-Fe2O3时臭氧分解的伪一级动力学拟合曲线. 根据拟合曲线斜率,得出加入α-Fe2O3后的分解速率常数明显大于单独臭氧. 因此,α-Fe2O3的加入既降低了溶解性臭氧平衡浓度,又加快了臭氧分解,这对于催化臭氧氧化过程是有利的.

图11 单独臭氧和催化臭氧的传质水平Fig.11 Mass transfer level of single ozone and catalytic ozone
2.5 ·OH屏蔽及界面反应探究
·OH的产生可能是有机物高去除率的主要原因,为进一步探究催化臭氧氧化过程中强氧化剂·OH的产生情况,开展了自由基屏蔽试验. 叔丁醇(TBA)是常用的自由基屏蔽剂,与·OH反应速率常数为6×108L/(mol·s). 在中性或酸性(pH≤7)条件下,TBA与臭氧几乎不反应. 图12(a)为反应过程中投加TBA后COD去除率随时间的变化曲线. 由图12(a)可知,反应60 min后,投加20和50 mg/L的TBA时,COD去除率较未投加时分别降低了7.59%、17.01%,这说明反应被明显抑制,催化臭氧氧化过程可能产生了·OH.为进一步证明·OH在催化过程的生成,采用DMPO作为·OH捕获剂,进行了EPR扫描〔见图12(b)〕. 结果表明,催化臭氧氧化体系表现出典型的峰强度比(1∶2∶2∶1). 但是,当单独使用臭氧和α-Fe2O3降解DMPO时,由于强度非常低,未观察到确定的峰值.由此可知,苯酚的高处理效率主要是由·OH的间接氧化造成的.
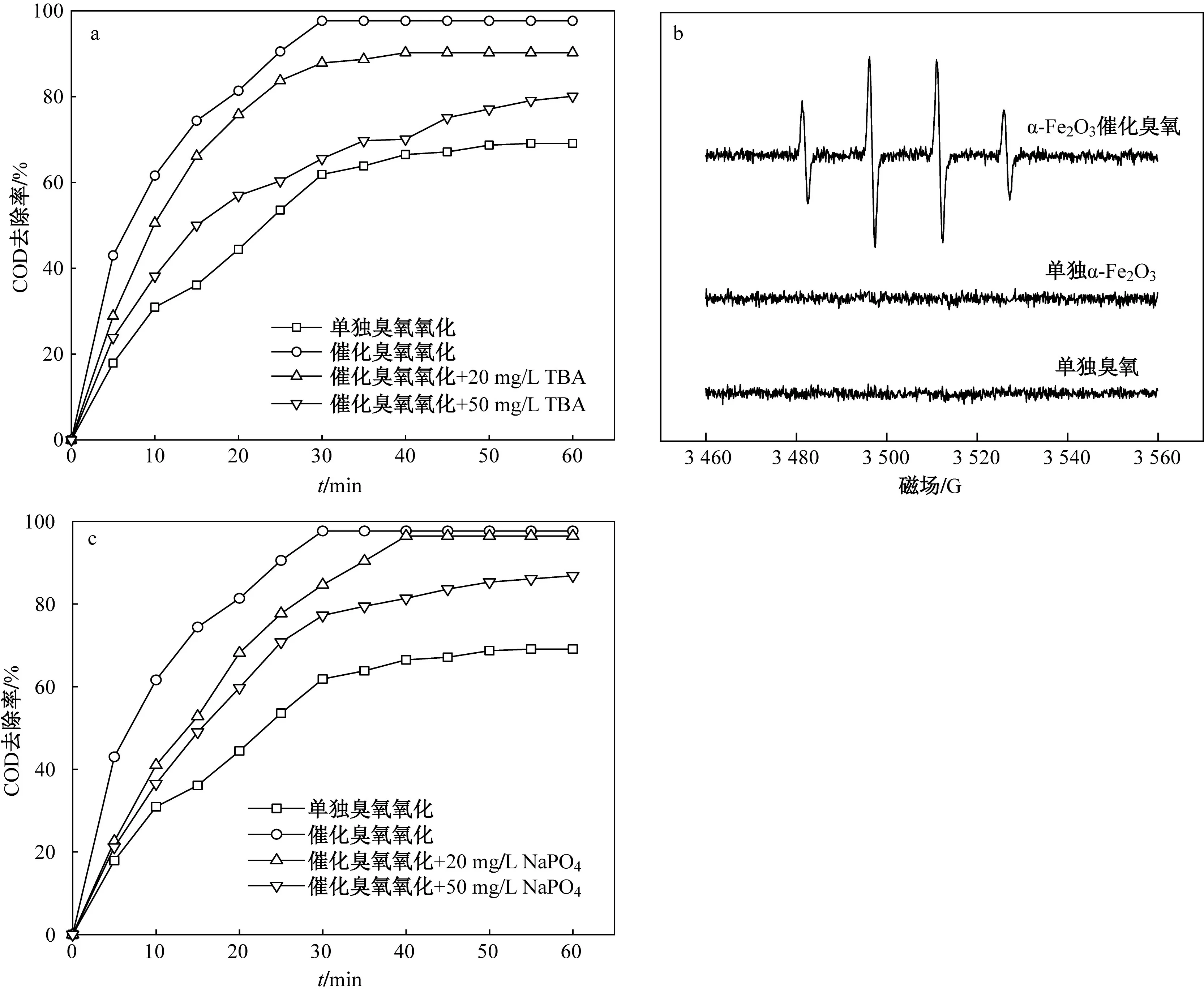
图12 ·OH捕获和反应位点试验Fig.12 ·OH capture and reaction site test
根据路易斯酸碱理论,金属氧化物存在Lewis酸性和碱性位点[35]. 其中,Lewis酸性位点源于催化剂表面的酸性羟基,能接受反应体系中含有的电子对,可与臭氧反应产生·OH,被认为是水相中催化臭氧氧化过程的反应中心[36]. PO43−、HPO42−、CO32−等无机阴离子常被用来研究催化剂的界面反应,能与催化剂表面Lewis酸性位点结合,可阻止臭氧分子接触,延缓·OH产生. 图12(c)为反应体系投加Na3PO4后COD去除率随时间的变化曲线. 由图12(c)可见,PO43−的加入延缓了反应速率,反应30 min后,投加20和50 mg/L的Na3PO4较未投加时分别降低了13.31%、20.92%,反应结束后,COD仍未被完全去除,这表明α-Fe2O3表面Lewis酸性位点是·OH重要的产生场所.
2.6 催化剂稳定性
催化剂稳定性是实际应用中关键的参数之一,有必要测定α-Fe2O3在循环使用中的催化活性及α-Fe2O3流失情况. 在最佳条件下对α-Fe2O3进行了6次重复使用,每次反应时间均为30 min,反应结束后补充苯酚和α-Fe2O3至初始浓度. 如图13所示,重复试验6次后COD的去除率从97.47%略微降至93.07%,仍保持了较高的去除效果. 然而,该过程性能轻微下降可能是由于反应活性位点的消耗和α-Fe2O3表面介孔阻塞. 溶液中Fe析出浓度随试验次数增加而逐渐升高,最终析出浓度为3.67 mg/L,α-Fe2O3流失率为1.05%. 综上,α-Fe2O3具有较好的催化臭氧氧化稳定性,可以成为去除有机废水经济且高效的催化剂.
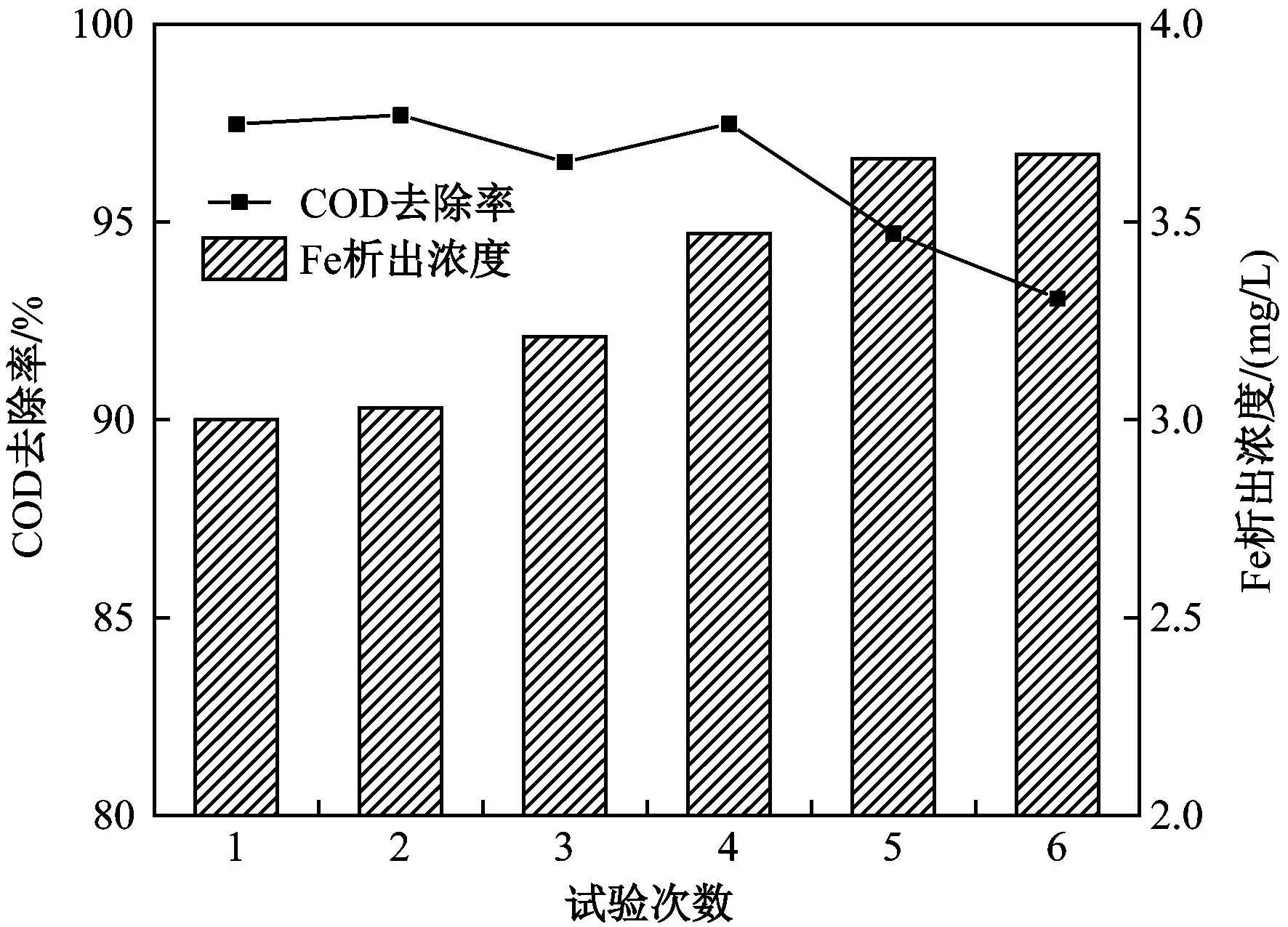
图13 重复试验中COD的去除率和Fe析出情况Fig.13 COD removal rate and precipitation of Fe element in repeated tests
3 结论
a) 通过对比α-Fe2O3、α-MnO2、TiO2、MgO、γ-Al2O3催化臭氧氧化去除苯酚废水COD的效果,证实α-Fe2O3具有更好的催化臭氧氧化效能. 在初始pH为7、臭氧投加量为10 mg/L、α-Fe2O3投加量为0.5 g/L时达到了最佳的苯酚废水COD去除效果,反应对弱酸性和弱碱性条件也具有一定适应性. 提高臭氧和α-Fe2O3投加量可提高苯酚废水COD的降解效率,但α-Fe2O3过量投加不利于反应进行.
b) α-Fe2O3高臭氧催化活性的主要原因是在反应过程产生了·OH,通过反应位点试验证实Lewis酸性位点是·OH的产生场所. α-Fe2O3在重复6次重复试验后保持了较高的催化活性和稳定性,COD去除率仍可达93.07%,流失率为1.05%. 采用α-Fe2O3催化臭氧氧化处理苯酚废水,具有较好的实际应用性.
猜你喜欢
今日农业(2022年4期)2022-11-16
工业安全与环保(2022年10期)2022-10-28
化学与生物工程(2022年9期)2022-09-30
环境工程技术学报(2022年3期)2022-06-05
能源化工(2021年6期)2021-12-30
煤气与热力(2021年10期)2021-12-02
煤化工(2021年5期)2021-11-24
科学大观园(2020年18期)2020-09-16
石油炼制与化工(2020年2期)2020-02-20
安全与环境工程(2018年3期)2018-05-31
