
医药中间体2,4,5-三氟-3-甲氧基苯甲酸的合成路线及应用研究进展
2022-08-16 09:38祁子丁蒋兴志雷旭松李德高
云南化工 2022年8期
田 强,祁子丁,蒋兴志,雷旭松,李德高
(1.云南云天化股份有限公司研发中心,云南 昆明 650600;2.云南云天化股份有限公司,云南 昆明 650600)
喹诺酮类抗菌药物具有结构简单、杀菌谱广、抗菌能力强、毒副作用小、给药方便等特点,成为近年来竞相开发与应用的热门药品。喹诺酮类属于化学合成类抗菌药物,自从1962年美国Lesher合成第一个喹诺酮类药物萘啶酸[1],翻开了化学合成抗感染药物新篇章;于1973年合成了以吡哌酸等为代表的第二代喹诺酮药[2];1978年合成了以诺氟沙星作为标志的第三代喹诺酮药[3]。目前已成为抗菌药物的的主流。由于第三代喹诺酮药结构中均含有氟原子,因此又称为氟喹诺酮类。
2,4,5-三氟-3-甲氧基苯甲酸化学分子式是:C8H5F3O3,其中文别名:3-甲氧基-2,4,5-三氟苯甲酸,英文名称:2,4,5-Trifluoro-3-Methoxy Benzoic Acid,是一种白色至淡黄色晶体粉末。主要用作医药中间体,是合成巴罗沙星(baloxacin)、加替沙星(gatifloxacin)等氟喹诺酮类广谱抗菌素的关键中间体[4]。
1 2,4,5-三氟-3-甲氧基苯甲酸的合成
目前,有关2,4,5-三氟-3-甲氧基苯甲酸合成工艺的研究有很多。本文按使用其初始原料的不同来对合成2,4,5-三氟-3-甲氧基苯甲酸的方法进行分类叙述。
1.1 以四氟邻苯二甲酰亚胺为原料
将四氟邻苯二甲酰亚胺按一定比例加入氢氧化钠溶液混合制得水溶液,加热溶液以产生4-甲氧基-3,5,6-三氟邻苯二甲酸盐和3-甲氧基-2,4,5-三氟苯甲酰胺的盐的混合物。用硫酸中和该溶液以形成4-甲氧基-3,5,6-三氟邻苯二甲酸和3-甲氧基-2,4,5-三氟苯甲酰胺的沉淀。使沉淀物与硫酸二甲酯反应以产生3-甲氧基-2,4,5-三氟苯甲酸[5]。合成路线见图1。

图1 以四氟邻苯二甲酰亚胺为原料的合成路线
1.2 以 2,3,4,5,6-五氟苯甲腈为原料
该法以 2, 3, 4, 5, 6-五氟苯甲腈为原料,先经过对位甲氧基化得到 2, 3, 4, 5, 6-五氟苯甲腈钠,再通过邻位氨化后,使用80%硫酸将其氰基转变酰胺基,再加入50%硫酸进一步水解为羧酸并脱羧得到3-甲氧基-2,4,5-三氟苯胺,再经过重氮化、氰化得到产品2,4,5-三氟-3-甲氧基苯甲酸[6]。合成路线见图2。
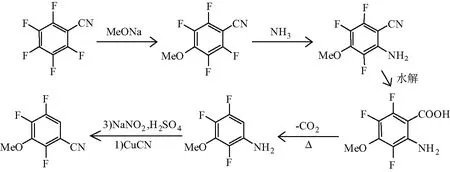
图2 以2, 3, 4, 5, 6-五氟苯甲腈为原料的合成路线
1.3 以2, 3, 5, 6-四氟-4-甲氧基硝基苯为原料
以 2, 3, 5, 6-四氟-4-甲氧基硝基苯作为起始原料,使用 Gabriel法先在其邻位上引入邻苯二甲酰亚胺基,再将其硝基还原为氨基后,再经过重氮化并脱氨基,然后在一定量的肼的存在下脱去其中的邻苯二甲酰基而得3-甲氧基-2,4,5-三氟苯胺,3-甲氧基-2,4,5-三氟苯胺再经重氮化、氰化得到产品2,4,5-三氟-3-甲氧基苯甲酸[7]。
1.4 以四氟邻苯二甲酸为原料
以四氟邻苯二甲酸为原料合成3-甲氧基-2,4,5-三氟苯甲酸路线有多条,目前国内大多数采用田治明所开发工艺路线:将四氟邻苯二甲酸加入25%的 NaOH水溶液中搅拌并加热,四氟邻苯二甲酸选择性地水解得到4-羟基-3,5,6-三氟邻苯二甲酸的钠盐,在保持体系呈碱性的条件下,向未经分离纯化的钠盐中加入少量盐酸以便中和部分碱,加热使其部分脱羧后再加入NaOH水溶液,使用 Me2SO4使其甲基化然后再用加入H2SO4酸化,即得产品2,4,5-三氟-3-甲氧基苯甲酸[8]。合成路线见图3。
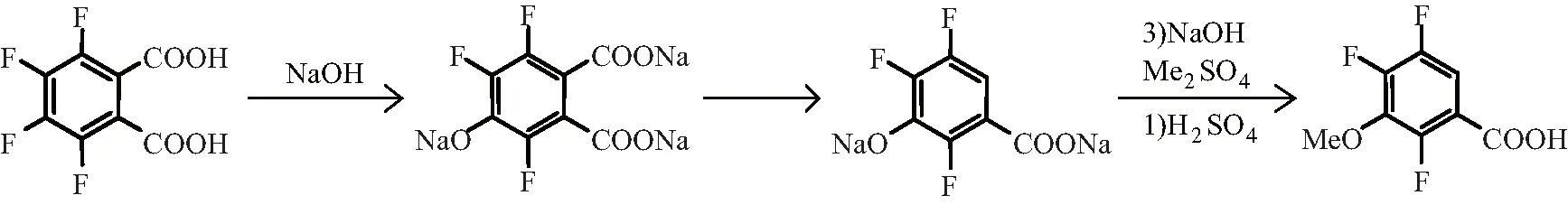
图3 以四氟邻苯二甲酸为原料的合成路线(纯碱法)
在2005年时俞伟梁发表了“一种合成2,4,5-三氟-3-甲氧基甲酸的方法”的专利,方法如下:先在回流温度和醇类溶剂中,在氢氧化钙和氢氧化钠混合碱体系中加入四氟邻苯二甲酸,进行脱氟甲氧基化从而获得中间产物2,4,5-氟3-甲氧基邻苯二甲酸;然后在二甲苯或甲苯溶液中和回流,2,4,5-氟3-甲氧基邻苯二甲酸和三正丁胺反应获得产品2,4,5-三氟-3-甲氧基苯甲酸[9]。合成路线见图4。
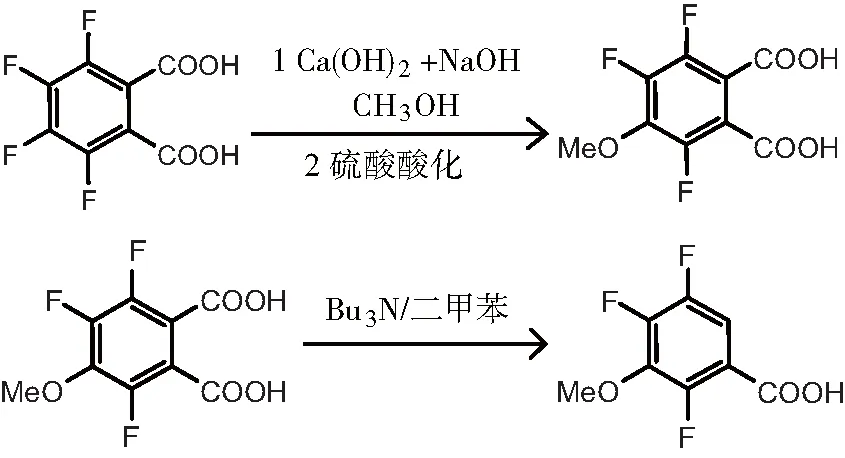
图4 以四氟邻苯二甲酸为原料的合成路线(混碱法)
1.5 以N-苯基四氯邻苯二甲化合物酰亚胺为原料
黄生建等以N-苯基四氯邻苯二甲化合物酰亚胺作为起始原料,以DMF为溶剂加入高活性氟化钾经过氟代反应,在强碱性环境下经过羟基化,再在水体系中加热至 120 ℃ 使其加压脱羧,然后加入甲醇中使其经过硫酸脱水成酯,所得酯化物在甲苯水体系中甲基化制得产品2,4,5-三氟-3-甲氧基苯甲酸[10]。合成路线见图5。
2 2,4,5-三氟-3-甲氧基苯甲酸的应用
2,4,5-三氟-3-甲氧基苯甲酸可以衍生出多种下游产品,主要用作医药中间体,是合成巴罗沙星(baloxacin)、加替沙星(gatifloxacin)等氟喹诺酮类广谱抗菌素的关键中间体。
2.1 合成加替沙星[11]
以2,4,5-三氟-3-甲氧基苯甲酸为起始原料,先经酰氯化、缩合、水解脱羧得2,4,5-三氟-3-甲氧基苯甲酰乙酸乙酯,然后再与原甲酸三乙酯进行缩合、烯胺化、环合得到中间体1-环丙基-6,7-二氯-1,4-二氢-8-甲氧基-4-氧代-3-喹啉羧酸乙酯,再经过水解反应,然后与2-甲基哌嗪缩合得到产品加替沙星(图5)。
加替沙星,为化学合成的氟喹诺酮类抗菌药,具有抗菌谱广的优点,尤其对革兰氏阳性菌有很强的抗菌活性。加替沙星最早于2002年在国内获得批准,主要用作治疗呼吸系统感染、泌尿系统感染以及由淋球菌感染引起的性传播疾病。其商品名包括:乐派、悦博、莱美清、奥维美、海超、罗欣严达等。
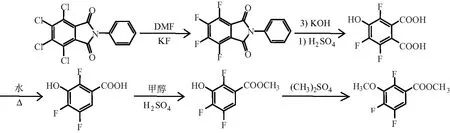
图5 以N-苯基四氯邻苯二甲化合物酰亚胺为原料的合成路线
2.2 合成巴洛沙星[12]
以2,4,5-三氟-3-甲氧基苯甲酸为起始原料,先经酰氯化、缩合(丙二酸乙二酯)、β-酮酯化、乙氧甲叉化,然后经过环丙胺置换及环化得中间体1-环丙基-6,7二-氟-1,4-二氢-8-甲氧基-4-氧代-3-喹啉羧酸乙酯,中间体在经过酰胺化后与3-甲氨基哌啶缩合制备得到1-环丙基-6-氟-1,4-二氢-8-甲氧基-7-(3-甲胺基-1-哌啶基)-4-氧代-3-喹啉甲酰胺,最后水解后得到产品巴洛沙星。
巴洛沙星适朱勇用于治疗非典型病原体引起的急性支气管炎、慢性支气管炎急性以及肺炎等呼吸系统感染;还可用于治疗前列腺炎、附睾炎、膀胱炎、肾盂肾炎、淋球菌性尿道炎等泌尿生殖系统感染;以及治疗沙门菌、细菌性痢疾及伤寒等引起的消化道感染;用于治疗皮肤软组织感染、骨感染、腹腔感染等疾病。巴洛沙星作为新一代喹诺酮类抗菌药物,其疗效可与第四代头孢类相媲美。
2.3 合成莫西沙星[13]
以2,4,5-三氟-3-甲氧基苯甲酸为起始原料,经过酰氯化后,在三乙胺溶液中与丙二酸单钾盐单乙酯发生缩合反应得到中间体(2,3,5-三氟-4-甲氧基)苯甲酰乙酸乙酯。在醋酐中β-酮酸酯与原甲酸乙酯回流,发生缩合反应生成3-乙氧基-2-(2,4,5-三氟-3-甲氧基)苯甲酰丙烯酸乙酯后,再与环丙基胺经加成-消除反应生成3- 环丙胺基-2-(2,4,5-三氟-3-甲氧基)苯甲酰丙烯酸乙酯,后者在DMF溶液中与氢氧化钠关环得到1-环丙基-6,7-二氟-8-甲氧基-1,4-二氢-4-氧代-3-喹啉羧酸乙酯,再在吡啶中用cis-(S,S)-2,8-二氮杂双环-[4.3.0]壬烷选择性取代其氟原子得到1-环丙基-7-{(S,S)-2,8-二氮杂双环[4.3.0]壬烷-8-基}-6-氟-8- 甲氧基-1,4-二氢-4-氧代-3-喹啉羧酸乙酯。最后,在浓盐酸和冰醋酸中回流水解得到产品莫西沙星。
莫西沙星属于第四代氟喹诺酮类抗菌药。莫西沙星对常见的呼吸道病菌,如嗜血流感杆菌、肺炎链球菌、卡他莫拉汉菌以及部分金黄色葡萄球菌均具有较强的抗菌活性。其作为DNA拓扑异构酶抑制剂,可用于治疗肺炎球菌、金葡菌、流感杆菌等引起的社会获得性肺炎,亦可用于治疗急性窦炎、慢性支气管炎急性发作等。
3 总结
喹诺酮类属化学合成的抗菌药物,具有抗菌谱广、抗菌力强、给药方便,疗效-价格比高等有点,近年来成为争相生产和应用的热点抗感染药品。全球喹诺酮类抗菌药物市场的快速增长,尤其是2000年以后,加替沙星(gatifloxacin),巴洛沙星(baloxacin)等抗菌药物经历了快速发展时期。对其重要中间体——2,4,5-三氟-3-甲氧基苯甲酸的需求量也在逐年增加,这为2,4,5-三氟-3-甲氧基苯甲酸的开展提供了一个充满活力的市场。因此,2,4,5-三氟-3-甲氧基苯甲酸合成工艺的研究对喹诺酮类抗菌药物的发展很有必要。紧密跟踪2,4,5-三氟-3-甲氧基苯甲酸的研究状况,并对现有的各种合成工艺路线进行深入的研究与分析,加强对其研究创新力度,必将会促使2,4,5-三氟-3-甲氧基苯甲酸的合成工艺水平不断完善和提升,这将进一步促进相关含氟精细化学品的蓬勃发展。
猜你喜欢
医药导报(2021年8期)2021-11-30
同位素(2021年5期)2021-10-23
安徽医药(2021年6期)2021-06-17
长江大学学报(自科版)(2021年3期)2021-06-01
农民致富之友(2018年12期)2018-06-29
饮食科学(2017年7期)2017-09-03
百科知识(2016年22期)2016-12-24
百科知识(2016年16期)2016-10-29
家庭百事通·健康一点通(2016年7期)2016-08-04
湖北畜牧兽医(2014年7期)2014-09-24
