
SOS1蛋白在肿瘤发生中的作用及其抑制剂研究进展
2022-08-16 08:43:56伍雪胡唯伟孙继红李雪杨勇
药学进展 2022年7期
伍雪,胡唯伟,孙继红,,李雪,杨勇*
(1.中国药科大学新药安全评价研究中心,江苏 南京 211198;2.南京圣和药业股份有限公司,江苏 南京 210018)
首个人类原癌基因RAS于1982年被Weinberg和Barbacid发现。后续研究证实细胞恶性转化与编码RAS蛋白的基因突变相关[1]。目前的研究发现,30%以上的人类肿瘤发生与RAS基因的突变相关[2-3]。RAS平滑的蛋白结构以及与三磷酸鸟苷(GTP)的高度亲和力使得RAS一度被人们称为“不可成药靶点”[4]。SOS1(Son of sevenless 1)作为RAS的上游蛋白,与RAS的活化相关。本文就SOS1在肿瘤发生中的作用及其抑制剂的研究进展进行综述。
1 RAS蛋白与肿瘤的发生
RAS基因在人类肿瘤细胞中易发生突变。目前已发现151种不同的RAS点突变,主要集中在G12、G13位以及Q61位,其中77%是G12位点的单突变[5]。
RAS蛋白作为下游通路的一种分子开关,依靠鸟嘌呤核苷酸交换因子(guanine nucleotideexchange factor,GEF)在不活跃状态(RAS-GDP)和活跃状态(RAS-GTP)之间循环转换[5]。RAS蛋白自身具有一定的GTP酶活性,GTP酶活化蛋白(GAP)帮助其促进GTP的水解。突变的RAS蛋白GAP活性显著降低,导致RAS永久激活[6]。
RAS通路的激活受信号分子的调控。信号分子[如表皮生长因子(EGF)]与酪氨酸激酶受体结合,导致后者的受体磷酸化。活化的酪氨酸激酶受体与生长因子受体结合蛋白2(Grb2)的SH2结构域结合。同时,Grb2的SH3结构域募集SOS蛋白形成复合物,将SOS蛋白定位于胞质膜[7]。定位于膜上的SOS蛋白催化RAS与GTP结合(见图1)。RAS-GTP可激活下游通路,其中最重要的是丝裂原活化蛋白激酶(MAPK)通路和磷脂酰肌醇3-激酶(PI3K)/蛋白激酶B(Akt)/哺乳动物雷帕霉素靶蛋白(mTOR)通路[8]。
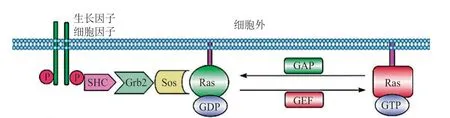
图1 RAS蛋白的激活过程[2]Figure 1 Process of RAS activation
三种RAS蛋白突变(包括KRAS、NRAS和HRAS这3种常见突变亚型)中,KRAS蛋白最易突变[2]。其与胰导管腺癌、大肠腺癌、肺癌的发生密切相关。KRAS基因编码KRAS4A和KRAS4B。KRAS4A正向调控肿瘤细胞的应激反应[3]。人类肿瘤中KRAS4B普遍存在且高表达[9]。肿瘤微环境中KRAS蛋白通过调节己糖激酶1(HK1)和乳酸脱氢酶A(LDHA)的表达来加快葡萄糖的摄取及能量代谢的过程,帮助肿瘤细胞更好地生存[10]。在KRAS突变中,已发现G12存在15种不同的点突变。其中,G12D突变频率最高,其次是G12V(约占28%)、G12C(约占14%)[11]。不同的KRAS突变与不同信号通路的激活相关,一般G12V和G12C突变与Ral GTPase(RAS信号传导的一个重要下游中介物[12])下游信号传导相关;G12D突变易激活PI3K通路[13],促进肿瘤发展。在白血病中,KRAS G12D的突变不仅可以激活下游通路,还可激活NRAS和HRAS的表达,加重病情[14]。
2 KRAS抑制剂耐药性的成因
目前,人们已发现KRAS抑制剂的耐药现象。其原因可归纳为以下两点。
1)产生旁路机制耐药。KRAS G12C抑制剂处理后可产生一部分新细胞。这些细胞保持在KRAS突变激活但对药物不敏感的状态[15-16]。有研究表明,这种耐药性与细胞的EGF受体(EGFR)和Aurora激酶信号转导相关[15],且EGFR过表达导致的受体酪氨酸激酶(RTK)的反馈激活与KRAS抑制剂MRTX849的耐药相关[17]。鼠类肉瘤病毒癌基因同源物B1(BRAF)受RAS调控,与丝裂原活化蛋白激酶激酶(MEK)的激活相关。但BRAF V600突变可独立激活MAPK通路,促进肿瘤生长[18-19]。抑癌基因NF1与PTEN的功能缺失突变可导致MAPK通路的过度激活[5]及PI3K/Akt通路的抑制解除[20]。在发生上皮间充质转化(EMT)的细胞中,PI3K/Akt通路也可由胰岛素样生长因子受体(IGFR)-胰岛素受体底物1(IRS1)通路激活,导致细胞对KRAS G12C抑制剂产生耐药现象[21]。局部黏着斑激酶(FAK)与肿瘤细胞的侵袭、转移及增殖相关[22],同时FAK也是KRAS下游的信号。有研究发现,KRAS G12C抑制剂可激活FAK,进而激活下游信号通路[23],使细胞出现耐药。
2)与组织学转化相关。肺癌的组织学分型转变可诱导肿瘤产生耐药现象[24]。在肺腺癌研究中发现,肺腺癌组织向鳞状细胞癌组织的转化促进细胞耐药[25]。
SOS1是RAS信号通路的上游节点。有研究发现,减少SOS1的表达或抑制其活性可降低KRAS突变细胞的增殖率及存活率[26]。因此,靶向SOS1的抑制剂不仅可用于治疗KRAS突变的肿瘤,还可克服KRAS抑制剂引发的耐药现象[27]。
3 SOS1蛋白的生物学功能及其在肿瘤发生中 的作用
3.1 SOS1的结构及其功能
SOS蛋白在人体中存在2个同源的亚基SOS1和SOS2[2]。这2个亚型具有不同的功能。在皮肤疾病方面,SOS1和SOS2都参与角化细胞的增殖和生存[28]。信号调节方面,SOS1优先参与细胞外调节蛋白激酶(ERK)的调节,SOS2主要参与PI3K信号的激活[28-29]。敲除SOS2基因的成年小鼠可以存活和繁殖[30],只是其毛囊中的干细胞数量显著减少[29],但敲除SOS1基因的小鼠发生胚胎致死现象[31]。因此,SOS1的生理学功能似乎更重要,大多数功能研究都集中于分析SOS1的功能。
SOS1蛋白具有多个结构域,其中包括HD、DH、PH、HL、REM、CDC25H以及PR[32-33]。其中,REM是变构位点,受RAS-GTP调控。CDC25H是催化位点,通过调整自身螺旋发卡的方向促进GDP向GTP的转换。REM、CDC25H以及PR共同参与RAS-GTP的形成。PH则与SOS1的膜定位相关。处于静息状态时,DH、PH和PR构成一个自身抑制模块[26],该模块可阻断REM结构域的结合位点并抑制CDC25H结构域的催化活性,从而抑制SOS1的生理学功能。当SOS1的REM结构域受到RASGTP的激活,其自身抑制状态解除,恢复GEF功能,这是SOS1受正反馈调节的激活机制[5]。有研究发现,SOS1 W729位点突变可破坏变构位点与RAS的结合[33];SOS1 F929位点突变可以破坏CDC25H结构域的催化活性以及影响SOS1的膜定位[33-34]。SOS1的膜定位对其生物学功能的发挥具有重要作用。帮助其定位的Grb2蛋白有2个结构域——nSH3和cSH3。这2个结构域分别与SOS1蛋白PR结构域中的PVPPPVPPRRRP和PKLPPKTYKREH多肽有高亲和力[34],破坏上述多肽将严重影响SOS1的生物活性。这一点在乳腺癌细胞的增殖中很关键[35]。SOS1 S1178位点也是SOS1膜定位的关键位点,该位点的磷酸化可阻断Grb2与SOS1的结合,使SOS1膜定位遭到破坏[36-38]。
3.2 SOS1蛋白与癌症
SOS1蛋白在RAS相关疾病(如努南综合征)中被大量关注[39],近来也有学者发现SOS1参与不同肿瘤的发展。
在肺腺癌中,SOS1突变与肿瘤的发生相关[14,32]。c-MYC基因是一种致癌基因,受MAPK通路信号的调控,并且c-MYC与胞内有氧糖酵解的过程相关[39],而有氧糖酵解释放的能量正是肿瘤细胞优先利用的能量。因此,c-MYC基因的表达与肿瘤细胞的增殖相关。突变的SOS1不仅可以上调RAS相关基因的表达、激活MAPK通路,还可以上调c-MYC的表达[32],促进肿瘤细胞的增殖。也有研究发现,未突变的SOS1蛋白高表达也可促进肺癌细胞在体外的增殖[40],这一点在急性髓系白血病细胞中也有体现[41]。
在胃癌中,SOS1可促进肿瘤细胞的恶性发展。这一过程主要与ErbB2/HER2信号通路的活化相关[42]。SOS1被RUNX相关转录因子1(RUNX1)反式激活,增强ErbB2/HER2信号通路的活化,促进胃癌细胞的恶性发展[42]。使用RUNX1抑制剂可有效抑制胃癌细胞的恶性发展。
在卵巢癌中,SOS1蛋白主要与肿瘤细胞的侵袭转移相关。SOS1蛋白与ABI1和EPS8形成复合物,该复合物是卵巢癌细胞转移的关键[43]。目前研究发现,TAT-p+p-8肽可通过抑制SOS1/EPS8/ABI复合物的形成,降低卵巢癌细胞侵袭转移的能力[43]。SOS1也可通过激活核因子κB(NF-κB)通路和Snail的转录诱导人卵巢癌HEY细胞骨架重排,促进细胞的侵袭转移[44]。SOS1不仅参与卵巢癌细胞的侵袭转移,也可通过促进c-Met通路的信号转导,参与乳腺癌细胞的生长、侵袭等过程[45]。
在胶质母细胞瘤中,SOS1蛋白的高表达不仅可以降低T98G细胞对miR-152-3p(一种miRNA)和顺铂联合用药的敏感程度,也与miR-152-3p治疗胶质母细胞瘤的脱敏性相关[46]。因此,检测SOS1蛋白的表达量在临床治疗中具有一定意义。
综上所述,SOS1蛋白的促瘤作用不仅与其激活MAPK及其下游通路相关,还与其自身蛋白水平的高表达相关。因此,靶向SOS1的药物既可以抑制RAS蛋白的激活,又可以抑制SOS1蛋白的功能,从而抑制肿瘤细胞的恶性进程。
4 靶向SOS1的小分子抑制剂
4.1 NSC-658497
NSC-658497是一种可阻断SOS1与RAS相互作用的先导抑制剂。在微摩尔级别呈剂量依赖地抑制SOS1GEF的活性。NSC-658497与SOS1的催化位点结合,阻断SOS1与RAS Switch II区域的相互作用。NSC-658497与SOS1结合的关键位点为I825、T828、T829和Y912。NSC-658497的芳香族苯并吡喃和极性硝基苯基是抑制SOS1活性的关键。NSC-658497不仅可以抑制突变型RAS细胞的增殖,同时还可抑制表达野生型KRAS的肺癌和卵巢癌细胞的生长[47]。作为早期的先导分子,研究者仅对NSC-658497进行了构效关系的研究,但目前尚未报道其在动物实验中的研究结果。
4.2 BAY-293
BAY-293是由拜耳公司通过高通量片段筛选得到的喹唑啉类抑制剂,其可破坏KRAS G12C与SOS1蛋白的结合(IC50= 21 nmol · L-1)。10 μmol · L-1BAY-293在KRAS突变的细胞系中,可抑制胞内50% p-ERK1/2的表达。在野生型的KRAS细胞中,MAPK通路也可被BAY-293抑制。在体外实验中,BAY-293与KRAS G12C抑制剂ARS-853可协同抑制肿瘤细胞的增殖。目前尚未查询到与该化合物相关的临床研究[48]。
4.3 BI-3406
BI-3406由勃林格殷格翰公司开发,是一种口服有效且具有选择性的SOS1抑制剂。与BAY-293相同,该化合物母核也是喹唑啉。喹唑啉环以π-π堆叠的方式与SOS1的His 905位点结合,甲氧基阻断SOS1 Tyr 884位点与KRAS Arg 73位点的结合。因此,BI-3406可阻断SOS1与KRAS的蛋白-蛋白相互作用。并且,BI-3406可抑制KRAS G12/G13/Q61突变细胞或野生型细胞内的p-ERK1/2(IC50为17 ~ 57 nmol · L-1),相比于Q61位点突变,G12和G13位点对BI-3406更敏感。然而,BI-3406并不能抑制野生型KRAS细胞的增殖;同时,一些KRAS突变且BRAF也突变的细胞(如HT-29细胞、A375细胞)对BI-3406的治疗也不敏感。因此,BI-3406单药治疗的效果是有限的,需要和其他抑制剂联用。有文献报道,BI-3406与MEK抑制剂联用,可增强肿瘤细胞对MEK抑制剂的敏感程度[5,49]。两药联用,可在体外显著抑制人胰腺癌MIA PaCa-2细胞(KRAS G12C)和人结肠癌DLD-1细胞(KRAS G13D)的增殖。在小鼠PDX模型中进行的体内研究也证明了同样的结果。并且,联合用药还可抑制小鼠体内原本对BI-3406不敏感的KRAS Q61突变细胞的增殖[5]。勃林格殷格翰公司并未申报BI-3406的临床研究,而是开展了其衍生物BI-1701963的临床研究。
4.4 BI-1701963
BI-1701963是首款进入Ⅰ期临床研究的SOS1抑制剂。BI-1701963通过与SOS1催化区域结合,可抑制SOS1与KRAS-GDP的结合,减少KRASGTP的形成,从而抑制MAPK信号通路的激活[50]。在结肠癌和胰腺癌的小鼠PDX模型中,BI-1701963与trametinib联用均可抑制KRAS G12V肿瘤生长。在结肠癌PDX模型中,BI-1701963与AMG510联用可显著抑制KRAS G12C肿瘤的生长,且联用效果优于AMG510或BI-1701963单独使用;BI-1701963与MRTX849联用可缩小KRAS G12C肿瘤体积,而单用MRTX849只能抑制该肿瘤的生长。BI-1701963与irinotecan联用可上调细胞内yH2AX蛋白(与DNA损伤相关)和Caspase 3蛋白的表达。在小鼠CDX模型中,BI-1701963与irinotecan联用对KRAS G12C、KRAS G12V及KRAS G13D肿瘤细胞都有显著的抑制效果;并且,联合用药还可缩小KRAS G12C和KRAS G12V肿瘤体积,而irinotecan或BI-1701963单独使用只能抑制肿瘤生长。在EGFR突变肿瘤中,有研究者发现奥希替尼与BI-1701963联用与奥希替尼单药相比,显著抑制MAPK通路和PI3K/AKT信号通路的激活[49]。
综上,BI-1701963不仅可以抑制KRAS突变肿瘤的增殖,也可增强肿瘤细胞对KRAS G12C抑制剂或irinotecan的敏感性。目前已开展多项临床试验,包括BI-1701963与trametinib联用(NCT04111458)、与BI-3011441联用(NCT04835714)、与MRTX849联用(NCT04975256)、与irinotecan联用(NCT04627142)和与BI-1823911联用(NCT04973163)治疗不同类型KRAS突变实体瘤的研究,除第一项研究外其余均处于招募阶段,目前尚未报道相关临床研究结果。
5 结语
SOS1是研究最充分的GEF,其在KRAS的激活中具有重要功能。目前在研的SOS1抑制剂大多数是小分子抑制剂,且结合靶点主要在His 905位点,药物分子通过占据这个位点阻断SOS1与KRAS的相互结合从而达到抑制KRAS激活的目的。目前SOS1抑制剂的临床研究报道甚少,即使是已进入临床的BI-1701963,其公开披露数据也不多,疗效和潜在毒性尚不明确。继续寻找高靶向性、低毒性的SOS1抑制剂是该类药物后续研究的重点;此外,基于RTK/RAS/MAPK通路复杂的调控机制,临床中单独使用SOS1抑制剂可能很难有效控制肿瘤的生长,探索联合用药的抗肿瘤效果也是未来研究的重要方向之一。
猜你喜欢
天津医科大学学报(2021年3期)2021-07-21 09:03:46
世界科学技术-中医药现代化(2021年12期)2021-04-19 12:31:40
广州大学学报(自然科学版)(2019年1期)2019-05-07 01:33:26
上海农业学报(2017年3期)2017-04-10 12:39:26
天津科技大学学报(2016年1期)2016-02-28 16:59:45
湖北师范大学学报(自然科学版)(2015年2期)2016-01-10 08:41:53
中国医药生物技术(2015年4期)2015-12-26 08:26:36
中国当代医药(2015年16期)2015-03-01 02:03:13
现代检验医学杂志(2015年2期)2015-02-06 02:01:01
中国药理学通报(2014年2期)2014-05-09 08:22:39
