
肿瘤源性IL-1β激活血管内皮细胞中NF-κB p65通路促进其对肝癌细胞的粘附*
2022-08-15 01:53房佩佩付晓燕吴沛恩吴圆圆张凯铖彭美玉梁淑娟
华中科技大学学报(医学版) 2022年3期
房佩佩,付晓燕,吴沛恩,吴圆圆,张 倩,张凯铖,彭美玉,梁淑娟
潍坊医学院免疫学教研室,山东省高校免疫学重点实验室,潍坊 261053
肿瘤微环境(tumor microenvironment,TME)是肿瘤转移过程中不可或缺的参与者,诸多研究表明炎症微环境促进肿瘤的发生和发展[1]。TME包括各类细胞成分和细胞分泌的可溶性细胞因子等,其中炎性细胞因子在维持炎症微环境和影响肿瘤细胞生物学行为中扮演了重要的角色[2]。肿瘤细胞占TME的主体成分,因此它们在TME中的作用也不容忽视。
肝癌(hepatoma)是一种典型的炎症相关肿瘤[3]。病毒感染、酗酒等产生的炎症刺激会导致肝癌的发生,而肿瘤形成后会通过各种机制继续在慢性炎症微环境的维系中发挥重要作用,例如分泌炎性因子肿瘤坏死因子α(tumor necrosis factor α,TNFα)、白细胞介素-6(interleukin-6,IL-6)、IL-8、IL-11[4-8]。一些对慢性炎症促发的肝癌模型的研究证实,肝癌细胞自身能通过产生IL-6而上调整合素表达,促进肝癌转移浸润[9],激活Kupffer细胞,招募和激活其他炎症细胞,从而维持炎症微环境[10]。饮食性或遗传性肥胖通过诱导肝细胞产生TNFα和IL-6而促进肝癌的发生发展[11]。IL-1β是一种重要的促炎性细胞因子,我们前期实验中证实过表达IL-1β可明显促进肝癌细胞浸润、转移,提示肿瘤源性IL-1β在加速肝癌浸润转移中发挥作用,但机制尚不完全清楚。鉴于肿瘤与血管内皮间的粘附是肿瘤浸润转移的关键环节,本实验着重探讨肿瘤源性IL-1β是否影响血管内皮对肿瘤细胞的粘附及其分子机制,重点分析了血管内皮细胞中E-选择素(E-selectin,即CD62E)、细胞间粘附分子-1(intercellular cell adhesion molecule-1,ICAM-1,即CD54)、血管细胞粘附分子-1(vascular cell adhesion molecule-1,VCAM-1,即CD106)的变化[12-14]及可能参与的信号机制。通过本研究期望对肝癌浸润转移提供有效的靶向和干预策略。
1 材料与方法
1.1 实验材料
载体pLKO.1-sp6-pgk-GFP、pLKO.1-sp6-pgk-IL-1β、psPAX2、pMD2.G,大肠埃希菌DH5α,HEK293T细胞,人肝癌细胞HepG2,保存于本实验室。qPCR引物由济南铂尚生物科技有限公司合成。质粒中提试剂盒购自Qiagen公司。RPMI 1640培养液及DMEM低糖培养液购自Gibco公司。新生胎牛血清购自Hyclone公司。LipofectamineTM2000、Human IL-1β ELISA试剂盒购自Invitrogen公司。Polybrene购自Santa Cruz公司。CFSE购自碧云天生物技术有限公司。PKH26购自MKBio公司,PMSF购自Merck公司。逆转录试剂盒购自Thermo公司。SYBR Green Ⅰ Master购自Roche公司。小鼠抗人CD62E-APC、CD54-FITC抗体购自Biolegend公司,FITC、APC同型对照购自BD Pharmingen公司。SDS-PAGE凝胶制备试剂盒购自Servicebio公司。细胞裂解液、蛋白酶/磷酸酶抑制剂Cocktail、兔抗人p38 MAPK、P-p38 MAPK、NF-κB p65、P-NF-κB p65、STAT3、P-STAT3 单抗及HRP标记山羊抗兔第二抗体购自Cell Signaling Technology公司。鲁米诺化学发光液购自Thermo公司。
1.2 实验方法
1.2.1 人IL-1β慢病毒表达载体的制备及感染人HepG2细胞 使用三质粒包装体系在HEK293T细胞中制备表达人IL-1β和对照GFP的慢病毒。将含有慢病毒的细胞培养上清在Beckman(SW32)超速离心机上4℃、25000 r/min离心2.5 h,将病毒浓缩10倍。在6孔培养板中接种1×105人肝癌细胞HepG2,加入相应的表达IL-1β和对照GFP的浓缩病毒100 μL/孔,感染4 d建立稳定表达IL-1β的细胞系(HepG2/IL-1β)和稳定表达GFP的细胞系(HepG2/GFP)。
1.2.2 双抗体夹心ELISA检测IL-1β浓度 收集6孔板中不同组别慢病毒感染48、72 h的HepG2细胞培养上清,4℃、12000 r/min离心10 min,取100 μL上清按照说明书检测IL-1β水平。含有IL-1β的肿瘤细胞条件性培养液(IL-1β-CM)作为肿瘤源性IL-1β的来源,以GFP慢病毒表达载体感染的HepG2细胞条件性培养液(Ctrl-CM)为对照。
1.2.3 细胞标记 2.5×104/孔人脐静脉内皮细胞(HUVECs)接种在48孔培养板中,按照说明书方法加入2 μmol/L PKH26标记后,将细胞置于37℃、5% CO2培养箱中培养过夜。按照说明书步骤,取1×106HepG2细胞加入2×CFSE储存液标记10 min。
1.2.4 细胞粘附实验 PKH26标记的HUVECs中加入含10 ng/mL IL-1β的IL-1β-CM或Ctrl-CM,刺激4 h或6 h。按照1∶1比例加入CFSE标记的HepG2/IL-1β或HepG2/GFP细胞2.5×104/孔。4℃静置孵育60 min,用PBS清洗3次,去除未粘附细胞。倒置荧光显微镜下观察并拍照、计数。共粘附率(%)=每100×视野下与HUVECs共粘附的肿瘤细胞数/肿瘤细胞数×100%。
1.2.5 qRT-PCR检测CD62E、CD54、CD106 mRNA水平 含10 ng/mL IL-1β的IL-1β-CM或Ctrl-CM刺激HUVECs 4 h,收集细胞,Trizol法提取细胞总RNA,测定RNA浓度及纯度。取4 μg总RNA进行逆转录,将逆转录产物cDNA 1∶4稀释,取1 μL进行qPCR,总反应体系为20 μL,以GAPDH为内参,通过2-ΔΔCt计算CD62E、CD54、CD106的mRNA相对表达水平。引物序列见表1。
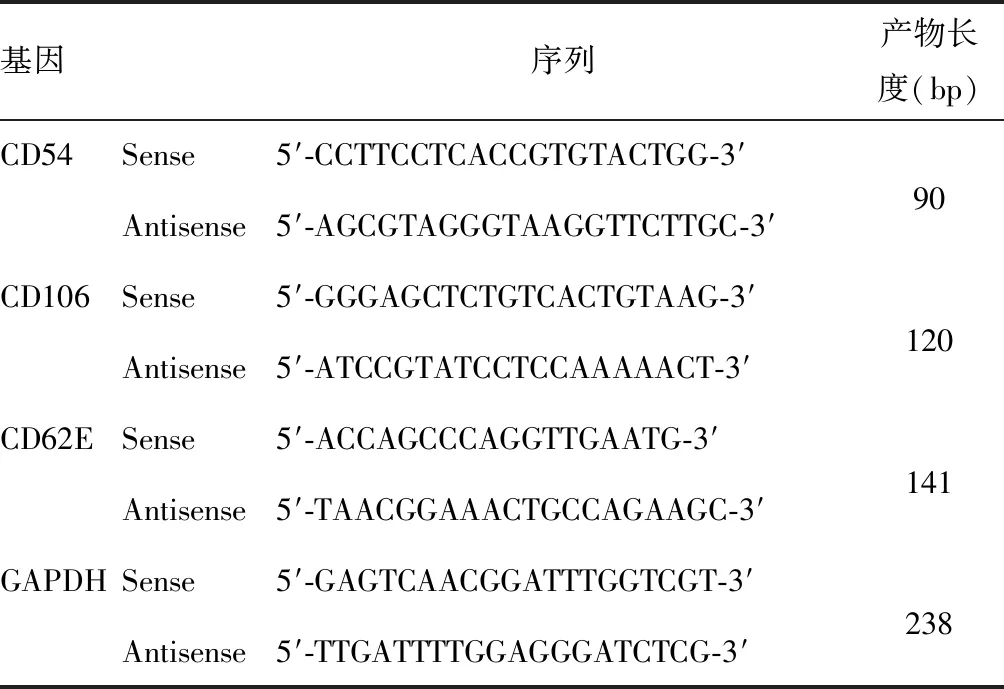
表1 qRT-PCR引物序列Table 1 Primer sequences of qRT-PCR
1.2.6 流式细胞术测定细胞表面粘附分子 含10 ng/mL IL-1β的IL-1β-CM或Ctrl-CM刺激HUVECs 4、6及24 h,收集1×106细胞进行分析。加入封闭抗体(anti-CD16/32)封闭30 min。加入anti-CD62E-APC、anti-CD54-FITC各5 μL/样本,4℃避光孵育30 min。FACS buffer洗涤2次,用300 μL FACS buffer重悬细胞,上机检测。
1.2.7 Western blot检测信号通路活化 HUVECs中加入含10 ng/mL IL-1β的IL-1β-CM,刺激时间分别为0、5、15、30、60 min,收集细胞,用于IL-1β对信号通路的时间依赖性激活分析。分析信号通路对IL-1β的剂量依赖性时,分别加入含0、0.1、1.0、10 ng/mL不同浓度IL-1β的IL-1β-CM刺激细胞5 min。每1×106细胞加入90 μL细胞裂解液,4℃裂解30 min提取总蛋白。蛋白定量后,取60 μg/样本总蛋白,进行10% SDS-PAGE电泳分离。结束后,将其转移至0.45 μm的PVDF膜上。加入5%脱脂奶粉溶液,室温封闭40 min。分别加入1∶1000稀释的兔抗人磷酸化和非磷酸化p38 MAPK、NF-κB p65、STAT3单克隆抗体,4℃孵育过夜。TBST洗膜3次,每次5 min。加入1∶2000稀释的HRP标记山羊抗兔第二抗体,室温孵育1 h。TBST洗膜5次,每次5 min,然后加入鲁米诺化学发光液,将其在AI600化学发光凝胶成像系统中曝光。
1.2.8 Western blot检测信号通路阻断效应 加入不同浓度的NF-κB p65通路特异性阻断剂Bay11-7082和p38 MAPK通路特异性阻断剂SB203580、SB202190阻断90 min,加入含10 ng/mL IL-1β的IL-1β-CM处理内皮细胞5 min,Western blot检测对信号通路活化的阻断效应。并观察对粘附分子表达、内皮细胞和肝癌细胞粘附作用的影响。
1.3 统计学方法
2 结果
2.1 慢病毒介导IL-1β在HepG2细胞中有效表达
慢病毒感染后,首先在6孔板中检测了培养不同时间时HepG2细胞培养上清中IL-1β水平。结果提示,自48 h起,稳定转染IL-1β慢病毒表达载体的HepG2/IL-1β细胞培养上清中IL-1β水平随培养时间延长不断升高。48 h时其浓度为100 ng/mL,72 h为150 ng/mL,均显著高于GFP对照病毒表达载体感染的HepG2/GFP细胞(图1)。鉴于慢病毒感染后48 h时HepG2/IL-1β细胞培养上清中IL-1β水平已经较高,且此时细胞状态良好,故后续实验中均以此时间点收获的IL-1β-CM作为肿瘤源性IL-1β的来源,并以HepG2/GFP细胞来源的条件培养液(Ctrl-CM)为对照。

**P<0.01图1 HepG2细胞培养上清中IL-1β的表达水平Fig.1 Expression level of IL-1β in HepG2 cell culture supernatant
2.2 肿瘤源性IL-1β增加HUVECs对HepG2细胞的粘附作用
为了确认肿瘤源性IL-1β是否增强血管内皮细胞对HepG2细胞的粘附作用,我们用IL-1β-CM或Ctrl-CM刺激血管内皮细胞HUVECs 6 h,然后分别加入HepG2/IL-1β、HepG2/GFP细胞进行4℃粘附实验。实验结果提示:含10 ng/mL IL-1β的IL-1β-CM刺激的HUVECs细胞组在培养皿中粘附的HepG2/IL-1β细胞总数明显多于HepG2/GFP细胞(图2A、2B);同时,与HepG2/IL-1β细胞的共粘附率显著高于HepG2/GFP细胞(图2C,P<0.01),提示IL-1β能够增加血管内皮细胞对HepG2细胞的粘附效应。在IL-1β-CM刺激内皮细胞HUVECs 4 h的粘附实验获得的结果与6 h实验呈相似趋势(图2D、2E)。含2 ng/mL IL-1β的IL-1β-CM刺激内皮细胞HUVECs进行粘附实验,HepG2细胞粘附的总数量和共粘附率比Ctrl-CM对照组细胞略有升高,但没有显著差异(P>0.05)。

A:肝癌细胞与HUVECs细胞(不同条件培养液刺激6 h)的粘附情况,白色箭头所示为共粘附,标尺=100 μm;B、C:肝癌细胞粘附数量及共粘附率(不同条件培养液刺激6 h);D、E:肝癌细胞粘附数量及共粘附率(不同条件培养液刺激4 h);*P<0.05,**P<0.01图2 4℃条件下IL-1β-CM(IL-1β 10 ng/mL)对肝癌细胞与血管内皮细胞粘附效应的影响Fig.2 Effect of IL-1β-CM(IL-1β 10 ng/mL)on the adhesion ability of tumor cells and vascular endothelial cells at 4℃
2.3 肿瘤源性IL-1β上调HUVECs中CD62E、CD54 mRNA和蛋白水平
与HUVECs+Ctrl-CM细胞相比,IL-1β-CM刺激后HUVECs细胞中CD62E(P<0.01)、CD54(P<0.01)分子mRNA的表达水平均明显升高,但是CD106 mRNA没有显著差异(图3A),故后续未再对CD106进行进一步观察。
流式细胞术分析细胞膜表面CD62E和CD54表达水平,结果显示,IL-1β-CM处理后HUVECs表面CD62E和CD54水平均显著升高,但其表达的动态变化有所差异(图3B~3E)。CD62E表达水平在IL-1β刺激4 h后迅速升高,平均荧光强度(MFI)明显高于Ctrl-CM处理的HUVECs,但是其表达升高仅维持短暂时间,6 h后水平下降,6 h和24 h时下降至与Ctrl-CM处理组持平的水平(图3B、3C)。而CD54在IL-1β刺激4 h后表达水平迅速升高,6 h时达到高峰,24 h略有下降,在此期间其MFI显著高于Ctrl-CM处理的HUVECs(图3D、3E)。

1:HUVECs+Ctrl-CM;2:HUVECs+IL-1β-CM;A:CD62E、CD54及CD106 mRNA的表达;B:动态检测CD62E蛋白表达的流式图;C:动态检测CD62E的平均荧光强度;D:动态检测CD54蛋白表达的流式图;E:动态检测CD54的平均荧光强度;*P<0.05,**P<0.01图3 肿瘤源性IL-1β对HUVECs中CD62E、CD54、CD106表达水平的影响Fig.3 Effect of tumor-derived IL-1β on the expression levels of CD62E,CD54 and CD106 in HUVECs
2.4 肿瘤源性IL-1β在HUVECs中主要激活NF-κB p65信号通路
Western blot结果提示,NF-κB p65、p38 MAPK两条信号通路的活化与IL-1β呈现时间和剂量依赖关系(图4、5)。IL-1β 10 ng/mL刺激HUVECs 5 min时,以上两条通路迅速激活,其活化可维持至30 min,60 min时活化程度下降(图4A、4 B)。而STAT3信号分子的磷酸化不随IL-1β刺激时间变化,提示其不被IL-1β激活(图4C)。1、10 ng/mL IL-1β对NF-κB p65、p38 MAPK通路的激活能力显著升高(图5A、5B),但STAT3活化水平不随IL-1β剂量变化。以上结果说明IL-1β主要是激活了HUVECs中NF-κB p65、p38 MAPK两条信号转导通路。为了进一步确认这两条信号通路是独立作用还是存在上下游依存关系,我们利用不同浓度的NF-κB p65通路阻断剂Bay11-7082及p38 MAPK通路阻断剂SB202190、SB203580分别进行阻断,观察它们对效应通路活化的影响。结果显示,40 μmol/L Bay11-7082显著阻断了NF-κB p65的磷酸化(P<0.01,图6A),同时p38 MAPK的磷酸化也有下降(P<0.01,图6B);40 μmol/L的SB202190或SB203580明显阻断了p38 MAPK的磷酸化(P<0.05,P<0.01,图6C),但对NF-κB p65磷酸化没有影响(图6D)。综上说明,NF-κB p65对p38 MAPK通路具有一定的调控作用,可能是IL-1β激发的主要信号机制。

A:NF-κB p65活化的时间依赖性变化;B:p38 MAPK活化的时间依赖性变化;C:STAT3活化的时间依赖性变化;*P<0.05 **P<0.01 图4 IL-1β作用不同时间对HUVECs中相关信号通路活化的影响Fig.4 Effect of different treating time of tumor-derived IL-1β on the activation of signaling pathways in HUVECs

A:NF-κB p65信号通路活化水平的剂量依赖性变化;B:p38 MAPK信号通路活化水平的剂量依赖性变化;C:STAT3信号通路活化水平的剂量依赖性变化;*P<0.05,**P<0.01图5 不同剂量IL-1β对HUVECs中相关信号通路活化的影响Fig.5 Effect of different doses of tumor-derived IL-1β on the activation of signaling pathways in HUVECs
2.5 阻断IL-1β相关信号通路下调HUVECs表面粘附分子表达并降低其对肿瘤细胞的粘附效应
利用Bay11-7082或(和)SB203580预先阻断90 min,分别观察其对IL-1β-CM诱导的HUVECs表面CD62E和CD54表达水平的影响。流式细胞术结果显示,单独使用Bay11-7082或联合SB203580均显著下调了IL-1β诱导的CD62E和CD54的表达水平(图7A、7B)。但是SB203580单独作用对以上两种粘附分子的表达无明显影响(图7A、7B)。这一结果提示,CD62E、CD54的表达主要受NF-κB p65信号通路的调控。
利用信号通路阻断剂阻断相应信号通路后,进一步观察了对HUVECs细胞与HepG2细胞粘附效应的影响。结果发现,预先在HUVECs细胞中将NF-κB p65阻断90 min后,显著降低了其对HepG2/IL-1β细胞的粘附作用(P<0.01),联合阻断p38 MAPK后,其阻断效应与单独阻断NF-κB较为接近(P<0.01);单纯阻断p38 MAPK通路,仅轻微影响肿瘤细胞的粘附作用(图7C、7E)。
3 讨论
大量研究证实,肝癌的发生发展和复发转移与肿瘤局部炎症微环境有着密切的关系,解析微环境中的关键调控机制有助于我们获取肿瘤生物学行为调控的重要靶标并为治疗提供有效干预策略。IL-1β是炎症微环境中一种重要的促炎细胞因子。正常生理条件下,IL-1β具有抗肿瘤保护机体的作用,而在慢性持续性炎症状态下IL-1β则能够促进肿瘤的发生发展[15-16]。IL-1β通过激活血管内皮生长因子(VEGF)促进肿瘤血管的生成[16]。IL-1β还可以诱导肺癌细胞ICAM-1的表达,促进肿瘤转移和侵袭[17]。在肝癌等肝脏疾病中,IL-1β在维系慢性炎症中发挥重要作用[18],IL-1β基因缺陷降低了肥胖诱发的肝癌发生率[19],
此外IL-1β通过上调Gankyrin和缺氧诱导因子1α(HIF-1α)促进肝癌的发生和转移[20-21]。但是IL-1β促进肝癌发展和转移的分子机制仍不完全明晰。本实验中,我们证实肝癌细胞来源的IL-1β能够显著促进血管内皮细胞与肿瘤细胞间的粘附作用,并对其机制进行了探讨。

11-7082对p65磷酸化的抑制作用;B:Bay11-7082对p38磷酸化的抑制作用;C:SB202190、SB203580对p38磷酸化的抑制作用;D:SB202190、SB203580对p65磷酸化的抑制作用;*P<0.05,**P<0.01图6 不同通路的特异性阻断剂对各信号通路活化的阻断效应Fig.6 Effect of different specific inhibitor on activation of NF-κB p65 and p38 MAPK pathways
肿瘤转移和肿瘤细胞与血管内皮细胞间的相互作用密切相关。实际上,肿瘤转移的过程与白细胞渗出过程类似。首先肿瘤细胞沿血管壁滚动,其表面的整合素激活,随即与内皮细胞发生粘附,启动穿越内皮的迁移过程[22]。炎症刺激下,血管内皮细胞表面粘附分子呈现动态表达:在刺激初始阶段血管内皮细胞表达CD62E,此期称为早期粘附,该过程中细胞间结合能力较弱,持续时间较短;之后发生晚期粘附,内皮细胞表达CD54和CD106,该过程中细胞间粘附牢固,持续时间长[23]。有研究发现IL-1β刺激HUVECs细胞4 h时CD62E的表达即增加[24]。CD54在血管内皮细胞低表达,TNFα和IL-1β刺激后,其在血管内皮细胞表达升高[25]。本实验动态检测了血管内皮细胞中上述分子在IL-1β刺激后的变化,结果发现,IL-1β-CM刺激4 h后CD62E分子表达增加,而6 h时其表达迅速下降,提示CD62E仅在肿瘤浸润内皮细胞的早期阶段发挥作用。与CD62E不同,CD54在IL-1β-CM刺激4 h至24 h,其表达水平均持续显著升高,提示CD54在介导肿瘤细胞与内皮细胞粘附中发挥重要作用。尽管有研究发现用TNFα分别处理人主动脉内皮细胞(HAEC)2 h和4 h时,能够上调CD106 mRNA和蛋白表达水平[26],LPS刺激HUVECs 24 h后CD106的表达也增加[27]。但是本研究中未发现IL-1β-CM对CD106 mRNA表达带来显著影响,推测其原因,一是由于不同的诱导因素可能对CD106的表达产生不同的效应,二是可能需要进一步优化检测的时间节点,后续研究中将对其表达情况进一步进行分析确认。
炎症因子通过激活细胞内相应信号机制发挥作用。IL-1β可以通过c-Jun途径上调HOXC10进而激活PDPK1和VASP表达促进肝癌转移[28]。在肝癌细胞中,敲除SPTBN1能促进NF-κB信号通路转录激活并上调促炎细胞因子IL-1α、IL-1β和IL-6释放,导致肝癌发生[29]。在肿瘤细胞中IL-1β还可以活化ERK1/2、p38 MAPK等通路[30]。有研究证实,在HUVECs中,IL-1β能够活化NF-κB、p38 MAPK信号通路[31],但是未对这两条通路的依存关系和对粘附分子表达的调控进行研究。本研究中,我们证实,肿瘤源性IL-1β通过激活NF-κB p65、p38 MAPK两条通路发挥作用,其中NF-κB p65位于p38 MAPK信号途径的上游,具有主控效应,是IL-1β在血管内皮细胞中活化的主要信号。NF-κB p65通路对内皮细胞表面CD62E和CD54的表达具有调控作用,并经由这两个粘附分子介导血管内皮细胞对肝癌细胞的粘附,靶向阻断NF-κB p65信号明显下调了粘附分子的表达和血管内皮细胞与肝癌细胞的粘附作用。
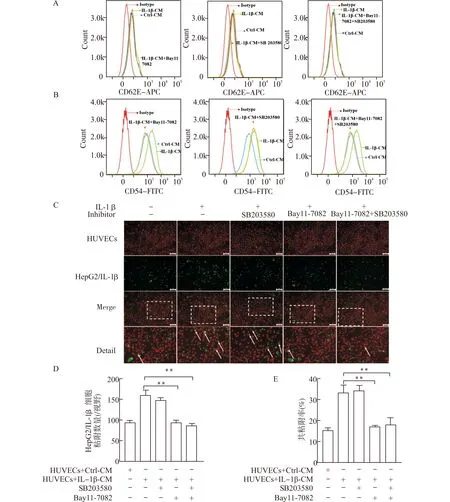
A:CD62E蛋白表达的流式图;B:CD54蛋白表达的流式图;C:HepG2/IL-1β细胞与HUVECs粘附情况,箭头所指为共粘附,标尺=100 μm;D:细胞粘附数量统计图;E:共粘附率统计图;**P<0.01图7 阻断信号通路后CD62E、CD54表达及HUVECs与肝癌细胞粘附情况Fig.7 The expression of CD62E,CD54 and the adhesion between hepatoma cells and HUVECs after blocking the signaling pathway
综上,我们的研究结果提示,靶向肿瘤源性IL-1β表达或靶向NF-κB p65可能成为干预肝癌浸润转移的新策略。但本文尚未对NF-κB p65下游更加精准的分子机制,例如关键性的转录因子等,进行深入探讨。另外,目前获得的主要是在体外研究取得的数据,后续尚需进一步在动物体内和临床样本中进行验证,以便获得更有价值的靶向策略。
猜你喜欢
康复(2022年25期)2022-11-18
基层中医药(2022年7期)2022-11-17
体育科技文献通报(2022年4期)2022-10-21
世界科学技术-中医药现代化(2022年3期)2022-08-22
现代仪器与医疗(2022年2期)2022-08-11
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
中国药理学通报(2022年1期)2022-01-14
现代临床医学(2021年5期)2021-11-02
分析化学(2017年12期)2017-12-25
