
Novel evolutionary insights into nemacheilid cavefish:evidence from comparative analysis of mitochondrial genomes*
2022-08-13 06:18LeiZHOUShihuiHUANGQingWANGZhenhaiLIZongyangLIAnyouHEJiehuCHENLiLIUKeshuZOU
Lei ZHOU , Shihui HUANG , Qing WANG , Zhenhai LI , Zongyang LI ,Anyou HE , Jiehu CHEN , Li LIU ,**, Keshu ZOU ,**
1 Joint Laboratory of Guangdong Province and Hong Kong Region on Marine Bioresource Conservation and Exploitation,College of Marine Sciences, South China Agricultural University, Guangzhou 510642, China
2 Guangxi Key Laboratory of Aquatic Genetic Breeding and Healthy Aquaculture, Guangxi Academy of Fishery Sciences,Nanning 530021, China
3 Science Corporation of Gene, Guangzhou 510000, China
Abstract Cavefish can be important models for understanding the relationships among evolution,adaptation, and development in extreme environments. However, cavefish remain poorly studied,particularly at the genome level. Here, we sequenced the complete mitogenome of three cavefish in the family Nemacheilidae ( Paranemachilus pingguoensis, Oreonectes polystigmus, and Heminoemacheilus longibarbatus), which were collected from karst caves in South China. The mitogenomes each contained 37 genes (13 protein coding, 22 tRNA, and two rRNA genes) and a single control region, with the same genetic arrangement and distribution as those found in vertebrates. The non-synonymous/synonymous mutation ratios (Ka/Ks) of the mitogenomes indicated that the protein-coding genes (PCGs) of the three cavefish evolved under purifying selection. The mitogenomes of the three cavefish exhibit nucleotide composition biases for PCGs, tRNAs, rRNAs, and the whole genome, indicating that the mitochondrial DNA might have been subjected to evolutionary selection in response to extreme cave environments. Divergence time and evolutionary history analyses suggested that the speciation and diversification of the cavefish coincided with the Miocene uplift of the southern Qinghai-Tibet Plateau, which greatly changed cave habitats. Overall, our study sheds light on the mitogenomes, phylogeny, and evolutionary history of nemacheilid cavefish.
Keyword: cavefish; mitochondrial genome; evolution; Nemacheilidae
1 INTRODUCTION
Cavefish are unique hypogean species whose evolutionary histories are associated with caves and other subterranean waters (Ma et al., 2019). There are two kinds of cavefish: stygobionts (also called typical or troglomorphic cavefishes) and stygophiles (also called atypical or non-troglomorphic cavefishes)(Zhao et al., 2011). Stygobionts are obligate cavefish whose adaptive traits, such as the degeneration or loss of eyes and reduced pigmentation, both enable and restrict them to complete their entire life cycles within cave environments. Stygophiles are not limited to hypogean aquatic environments but rely heavily on cave habitats during their life cycles (Ma et al., 2019).Globally, more than 350 species of cavefish live in caves and other subterranean habitats, including at least 199 species of stygobionts (Niemiller et al.,2019). Most cavefish species are found in the subtropics and tropics, and 75% of known populations are located in Southeast Asia or Central and South America within karst landscapes (Ma et al., 2019).
There are 148 described species of cavefish in China (accounting for 39% of such species worldwide),most of which reside within the 620 000 km2of karst landscape in the southwest (Huang et al., 2008). Karst landscapes feature complex surfaces and underground systems and provide diverse habitat to aquatic and terrestrial species (Deharveng et al., 2009; Luo et al.,2016). However, most species are limited to one independent cave or subterranean stream (Shu et al.,2013). For example,Heminoemacheilushyalinusis only distributed in a single cave in Du’an in the Guangxi Zhuang Autonomous Region. Karst habitats comprise variable and unique geological structures that can harbor communities of highly endemic and endangered species (Luo et al., 2016); these habitats are extremely fragile and sensitive to environmental change (Zhao et al., 2011; Palandačić et al., 2012).Caves are natural laboratories for ecological and evolutionary research, and the ability of cavefish to survive in extreme environments makes them excellent models for adaptation studies (Riddle et al., 2018).However, diffi culties in accessing and sampling remote caves have limited study of these ecosystems(Shu et al., 2013). As such, many cave species remain poorly studied, particularly with regard to their genome-level characteristics.
Cave environments are characterized by permanent darkness. The lack of autotrophic primary producers in dark environments can lead to restrictions in food supply; hence, cavefish have likely developed strategies to enhance their energy utilization to survive. Mitochondria play vital roles in energy metabolism. Cavefish mitochondrial DNA (mtDNA)may have undergone positive directional selection in response to the extreme environmental pressures in cave habitats (Boore, 1999; Xu et al., 2007; Tomasco and Lessa, 2011). Generally, mtDNA is characterized by its simple structure, small size, maternal inheritance, and rapid evolution (Avise et al., 1984)and has been extensively studied in relation to taxonomy, genetics, phylogenetics, and evolution(Satoh et al., 2016; Liu et al., 2017). However, few fully sequenced cavefish mitochondrial genomes(mitogenomes) are available with which to study adaptive evolution to subterranean life.
Nemacheilidae is the most diverse cavefish family(53 species) (Niemiller et al., 2019), which may have arisen due to the strong selective pressures found in isolated cave habitats (Zhao and Zhang, 2009). In southwest China, nemacheilid cavefish exhibit rich species diversity and some species, such asParanemachiluspingguoensis, have relatively large population sizes (Lan et al., 2013). Nemacheilid fish can be valuable for systematic study of cavefish evolution and adaptation and can serve as references for the study of karst development in the Yunnan-Guizhou area. However, despite their great scientific value, Nemacheilidae cavefish have been much less studied than their surface-dwelling counterparts, with very few studies to date (Lan et al., 2013).
In the current research, the complete mitogenomes of three nemacheilid cavefish species, i.e.,Paranemachiluspingguoensis,Oreonectespolystigmus,andHeminoemacheiluslongibarbatus, were sequenced. In morphology,O.polystigmuscan be distinguished fromP.pingguoensisandH.longibarbatusby caudal fin rounded and tip of anterior nostril elongated and barbel-like.H.longibarbatusandP.pingguoensisshare closely set nostrils but can be separated from each other by cheek scales present inParanemachilusvs. absent inHeminoemacheilus. The genomic characteristics of these cavefish were compared with those of closely related species within Nemacheilidae to investigate genetic adaptations to cave habitats. The evolutionary history of the nemacheilid cavefish was explored and found to be linked to geological events that altered the karst environments.
2 MATERIAL AND METHOD
2.1 Sample collection
Paranemachiluspingguoensis(tag number: PG-20190701; GPS: 23.33°N, 107.32°E),Oreonectespolystigmus(tag number: DB-20190701; GPS:25.03°N, 110.31°E), andHeminoemacheiluslongibarbatus(tag number: CX-20190701; GPS: 24.11°N,108.10°E) specimens used in this study were collected in Guangxi Zhuang Autonomous Region, China in July 2019 using a cage net (mesh size: 0.70 cm,length: 20 m, width: 45 cm, height: 33 cm). After collection, specimens were euthanized in tricaine mesylate (MS-222), and then dissected. Muscle tissues were preserved in 95% ethanol in the field and transferred to a -80-°C freezer for long-term storage in a laboratory at the College of Marine Sciences,South China Agricultural University (SCAU), China.The experiments were approved by the Animal Ethics Committee of SCAU (SYXK-2019-0136).
2.2 DNA extraction and mitogenome sequencing
Genomic DNA was extracted from the muscle tissue samples using an EZNA®Tissue DNA kit(Omega Bio-tek, USA) following the manufacturer’s instructions. The DNA was evaluated and quantified using 1.0% gel electrophoresis followed by ultraviolet spectroscopy (NanoDrop 2000, Thermo Fisher Scientific, USA). The resulting high-quality genomic DNA was isolated and used for library construction(TruseqTM RNA Sample Prep Kit, Illumina, USA),with a 350-bp insert used for paired-end (150 bp)sequencing on the HiSeq4000 platform (Illumina).The mitogenomes then underwent next-generation sequencing following the manufacturer’s instructions.
2.3 Analysis of sequence data
Raw sequencing data were retrieved and trimmed using Cutadapt v1.16 software with the parameters -q 20 -m 20 (http://cutadapt.readthedocs.io/) to obtain clean data. The clean paired-end reads were then used for de novo assembly using NOVOPlasty v3.8 software (Dierckxsens et al., 2017).
The mitogenomes were annotated using Mitofish v3.51 (Iwasaki et al., 2013) and ORF Finder (https://www.ncbi.nlm.nih.gov/orfinder/). The preliminary results were compared with the coding proteins and rRNA of mitogenomes from related species, using Blastp and Blastn (https://blast.ncbi.nlm.nih.gov/Blast.cgi), respectively, to confirm and correct the accuracy of the results. The complete mitochondrial DNA sequences were deposited in GenBank under accession numbers MW043720 (P.pingguoensis),MW043721 (H.longibarbatus), and MW043722(O.polystigmus).
Mitochondrial codon usage biases can aff ect gene expression and reflect the evolutionary relationships among species (Wei et al., 2014). Therefore, the relative synonymous codon usage (RSCU) for each genome was analysed (Sharp and Li, 1987). Repeat sequences were isolated using Tandem Repeats Finder v4.04. The skew associated with nucleotide bases was calculated using the following formulae: adenine (A)and thymine (T) skew=(A- T)/(A+T) and guanine (G)and cytosine (C) skew=(G- C)/(G+C). Nonsynonymous/synonymous mutation (Ka/Ks) ratios were calculated and applied to investigate the influence of selective pressures on protein-coding genes (PCGs) in cavefishes and related species; this was performed using the seqinr package in R software(Charif and Lobry, 2007).
2.4 Phylogenetic analysis
The mitogenome sequences of 61 nemacheilid species were subjected to phylogenetic analyses, withLeptobotiapellegrini(NC_031602) rooted as the outgroup. We aligned the individual genes for all species using MUSCLE v.3.8.31 (Edgar, 2004)according to the selected coding sequence. Following alignment, the genes of each species were then connected into a series that was integrated into a corresponding PCG sequence set for the following analyses.
Phylogenetic analyses were conducted using two approaches: maximum likelihood (ML) and Bayesian inference (BI). The DNA models were tested using jModelTest (Posada, 2008) based on selected sequences; the best model was selected based on the minimum Akaike information criterion. For the ML phylogenetic analysis, RAxML v8.1.5 software(Stamatakis, 2014) was applied using 1 000 bootstrap replicates. The number of replicates was determined automatically using the protein substitution model with the highest score. For the BI phylogenetic analysis, MrBayes v3.2.6 (Ronquist et al., 2012) was used, and results were considered reliable if the split frequency standard deviations were <0.01.
A molecular clock was constructed using MEGA X v10.1.7 software (Kumar et al., 2018) following phylogenetic and corresponding sequence analyses using ML. The diff erentiation times among species were queried using TimeTree (Kumar et al., 2017)with the following reference diff erentiation times:11.70 million years ago (Ma)- 45.42 Ma forTriplophysastenuraandSchisturafasciolata, 1.50 Ma- 11.69 Ma forLefuanikkonisheandLefuacostataand 48 Ma-103 Ma forOreonectesplatycephalusandTriplophysastenura.
3 RESULT
3.1 Mitogenome organization and composition
The complete mitogenomes ofOreonectespolystigmus,Paranemachiluspingguoensis, andH.longibarbatuswere 16 568, 16 562, and 16 569 bp in length, respectively. They each contained the canonical 13 PCGs, two rRNA genes (12Sand16S),22 tRNA genes, and a D-loop control region. The majority of the genes are encoded by the heavy strand(H-strand), except for nicotinamide adenine dinucleotide hydride (NADH) dehydrogenase subunit 6 (ND6) and eighttRNAgenes for glutamine, alanine,asparagine, cysteine, tyrosine, serine, glutamate, and proline, which were all located on the light strand(L-strand) (Fig.1 and Table 1). The two rRNAs,16Sand 12S, were located betweentRNA- Leu ( TAA) andtRNA- Val ( TAC) and betweentRNA- Val ( TAC) andtRNA- Phe ( CAA), respectively (Fig.1).
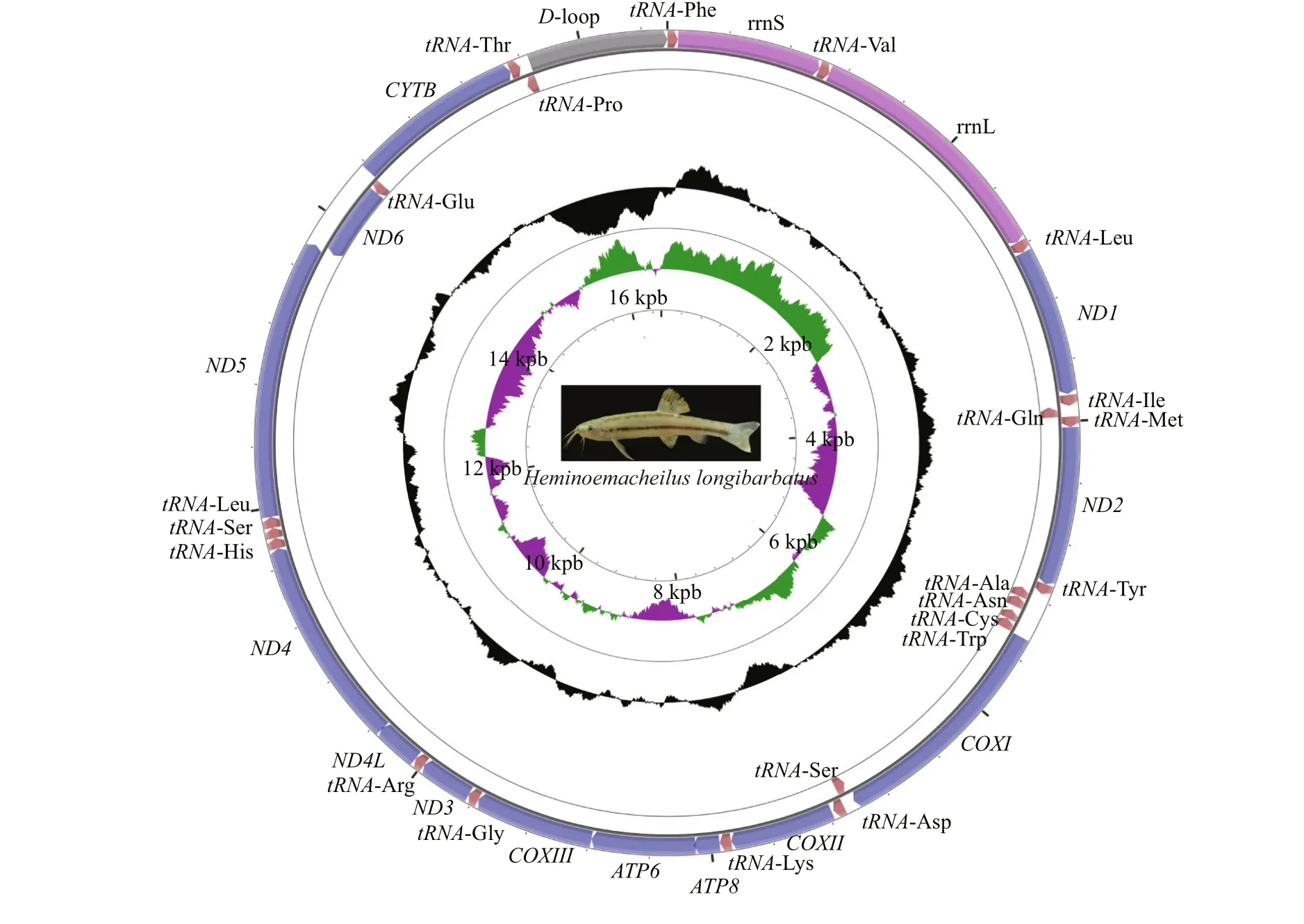
Fig.1 Continued
3.2 Base composition and base bias
The nucleotide composition (H-strand) of the three cavefish in descending order was 30.13% A, 27.69%C, 25.75% T, and 16.43% G, with an AT content of 55.88% (54.91%-56.44%).
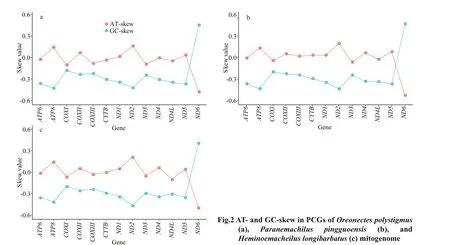
Most PCGs had a negative GC skew, except forND6, which exhibited a positive GC skew (Fig.2).Furthermore, the AT skews of most PCGs were around zero, except forND6, which had a negative AT skew(-0.48, -0.53, and -0.50 forOreonectespolystigmus,Paranemachiluspingguoensis, andHeminoemacheilus longibarbatus, respectively).
3.3 PCGs and codon usage
The coding regions of the three species were 11 427 bp (3 800 amino acids) in length, accounting for approximately 69% of the total mitogenome. The initiation codon for most PCGs was the canonical methionine sequence (ATG), with the exception of the start codon forCOXI, which was GTG. We also identified T, TA, TAA, and TAG termination codons in the cavefish mitogenomes, with TAA and T being the most prevalent. The TAG termination codon was only found in theND1,ND5, andND6genes ofHeminoemacheiluslongibarbatus(Table 1).
For the three examined species, all amino acids were encoded by two or four codon variations, except for leucine and serine, which were encoded by six(Fig.3). RSCU analysis revealed that the following codon sequences had higher frequencies: stop (TAA),arginine (CGA), and leucine (CTA). According to RSCU values, codons with A or C in the third position occurred more frequently than those with T or G (Fig.3).
The PCG Ka/Ks ratios were all <1 (Fig.4). The Ka/Ks ratios forATP8andND2were the highest (Fig.4).
3.4 Genes encoding tRNAs and rRNAs
In total, 22 tRNA genes were identified in the three cavefishes, ranging from 66 bp (tRNA- Cys) to 75 bp(tRNA- Leu andtRNA- Lys) in length. In general,codons and anticodons exhibited a one-to-one relationship. However, two anticodons were present for both serine (TGA and GCT) and leucine (TAG and TAA). The dihydrouridine (DHU) arm oftRNA- Ser(GCT) was absent, whereas the secondary structures of the other 21 tRNAs exhibited typical clover structures.
3.5 Compositional biases between cavefishes and other nemacheilid species
The genomic sequences of other cavefishes(stygobionts and stygophiles) and other nemacheilid species were compared to investigate diff erences in nucleotide composition. Absolute value of GC and AT skews were higher in the cavefishes than in other nemacheilid species (Fig.5); this was the case for the entire mitogenome, as well as for individual elements such as PCGs, rRNAs, and tRNAs.Principal component analyses (PCA) suggested that the overall codon and amino acid usage patterns of cavefishes were notably diff erent from those of other fish (Fig.5e & f).
3.6 Phylogenetic and divergence time analyses of cavefishes
A phylogenetic tree was constructed based on the mitogenomic PCGs of 61 nemacheilid species,including three species of stygobionts and five species of stygophiles, withLeptobotiapellegrinias the outgroup (Fig.6). Both ML and BI approaches produced trees with similar topologies and branch lengths, indicating a well-resolved and highly supported phylogeny of Nemacheilidae. The results show that the 61 nemacheilid species could be divided into four branches or clades. Clade I included all species ofTriplophysaandBarbatula. Clade II was sister to Clade I and comprisedHomatula,Schistura,andTuberoschistura. Clade III included the generaHeminoeacheilus,Micronemacheilus,Oreonectes,Troglonectes,Paranemachilus,Lefua, andTraccatichthys. The other eight genera,Petruichthys,Schistura,Nemaceilus,Oxynoemacheilus,Nemachilichthys,Hedinichthys,Acanthocobitis, andAborichthys, were clustered into Clade IV, which was positioned at the base of the Clade III + (Clade II +Clade I) clade.
The cavefishes were distributed across five genera(Triplophysa,Heminoemacheilus,Oreonectes,Troglonectes, andParanemachilus) in Clade I and Clade III. In Clade I, the genusTriplophysaappeared to diverge from the other species around 24.83 Ma(Fig.6). InTriplohysa, the divergence of two stygobionts(TriplophysaxiangxiensisandTriplophysarosa) from other non-cave species was estimated to have occurred around 20.93 Ma during the Miocene epoch. In Clade III, the cavefishes (Paranemachilusjinxiensis,Paranemachiluspingguoensis,Troglonectes daqikongensis, andTroglonectesfurcocaudalis)diverged from their sister groupLefuaaround 24.65 Ma during the Chattian age of the late Oligocene epoch. The stygophileOreonectespolystigmusdiverged from its surface counterpart (Oreonectes platycephalus) circa 6.40 Ma during the Messinian age of the late Miocene epoch, and the stygophileHeminoemacheiluslongibarbatusdiverged from its surface counterpart (Y.cruciatus) circa 18.91 Ma during the Burdigalian age of the Miocene epoch. In terms of divergence times among cavefishes, the occurrence range was 7.71 Ma (TriplophysaxiangxiensisandTriplophysarosa) to 13.76 Ma (TroglonectesdaqikongenisandTroglonectesfurcocaudalis)(Serravallian to Messinian Age, Miocene Epoch).
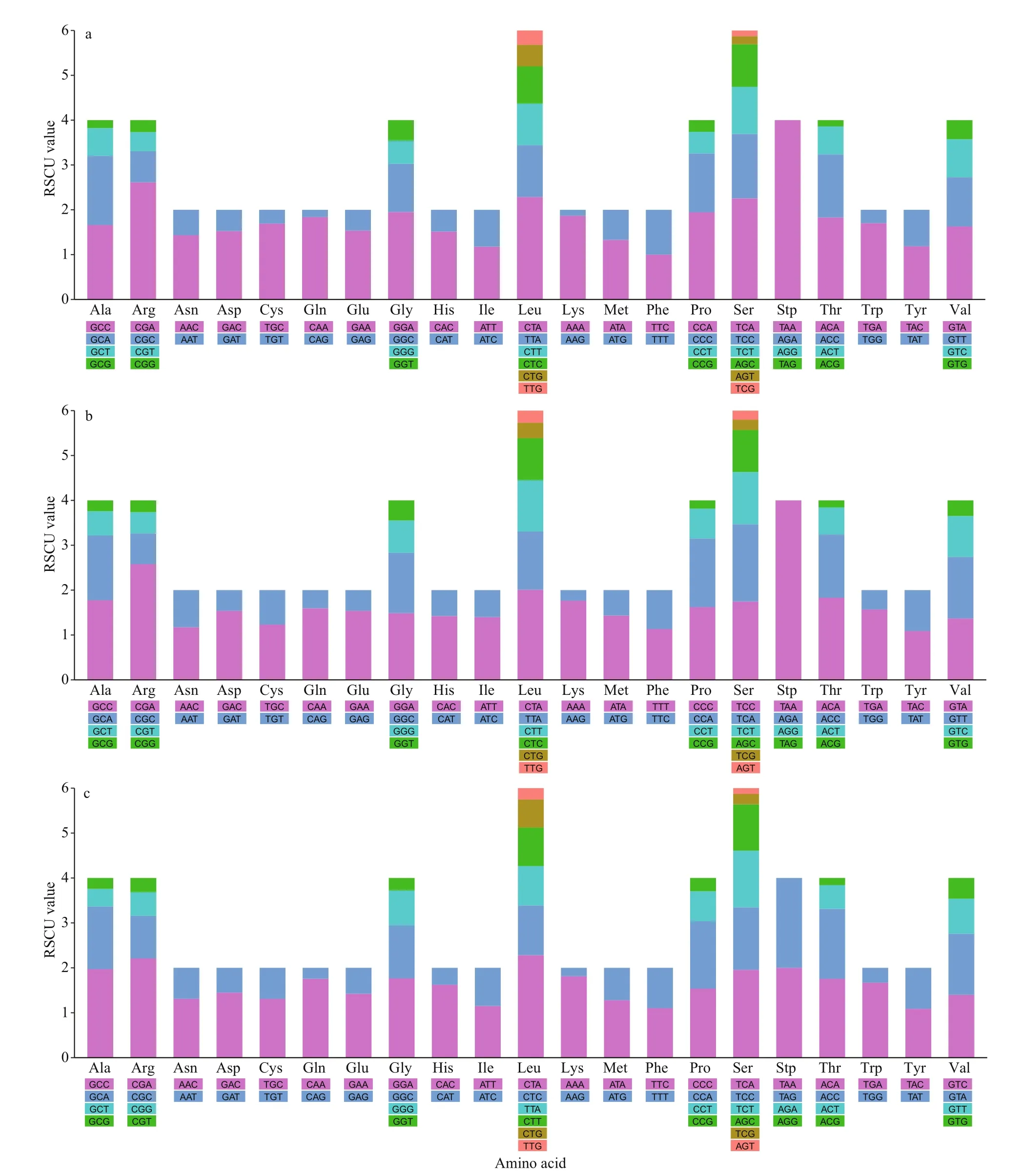
Fig.3 Relative synonymous codon usage (RSCU) of mitogenomes for Paranemachilus pingguoensis (a), Oreonectes polystigmus (b), and Heminoemacheilus longibarbatus (c)
4 DISCUSSION
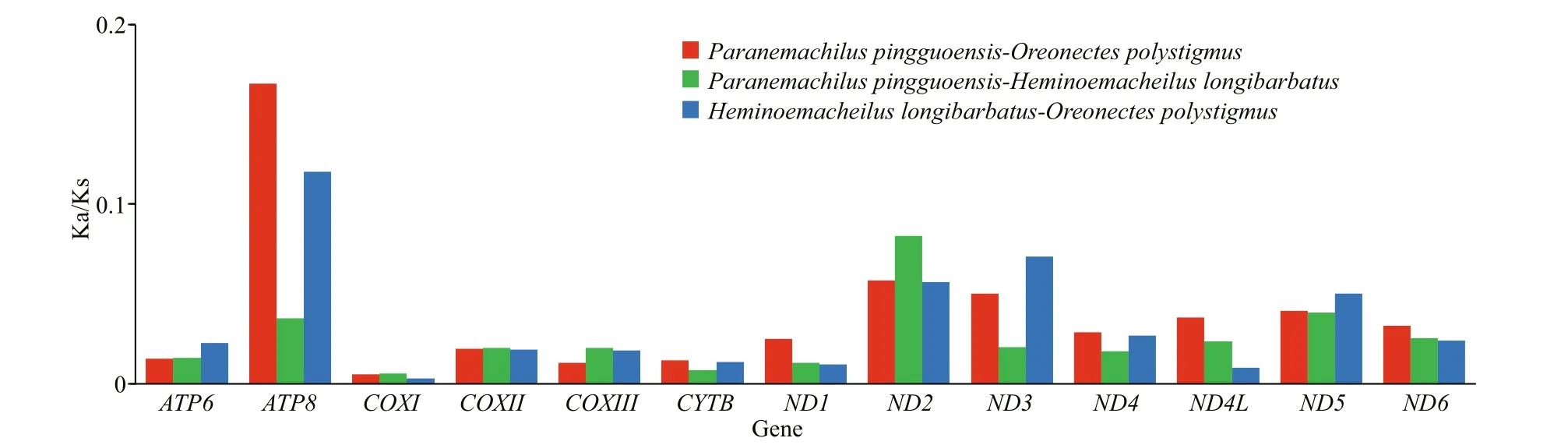
Fig.4 Ka/Ks ratios of 13 protein-coding genes in pairwise mitonchondrial genome of Paranemachilus pingguoensis,Oreonectes polystigmus, and Heminoemacheilus longibarbatus
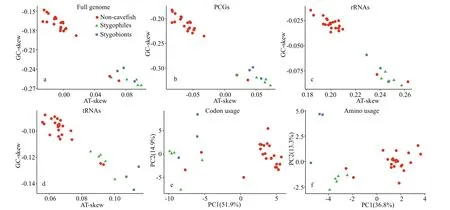
Fig.5 Comparison of nucleotide skews, codon usage, amino acid usage of the mitogenomes of stygobionts, stygophiles, and other Nemacheilidae species
Previous research has shown that certain teleostean fish possess only one or similar arrangements of mitochondrial genes (Miya et al., 2003). We found similar gene arrangements in the mitogenomes of the three cavefish species, which are also teleostean fish.Gene arrangement and distribution without subsequent gene rearrangement is a common feature in vertebrates(Broughton et al., 2001). The genomes of all examined nemacheilid species exhibited higher AT content relative to GC, consistent with the high AT and low GC contents displayed in most vertebrates (Cheng et al., 2012; Luo et al., 2017; Kundu et al., 2018; Huang et al., 2019). However, negative AT skews were found inND6in the three cavefish, which are uncommon in mitochondrial genes, but have been detected in other vertebrates such asMarmotahimalayana(Chao et al.,2014),Cetonurusglobiceps(Shi et al., 2016), andOphichthusbrevicaudatus(Lü et al., 2019). These animals inhabit extreme environments, a feature shared by nemacheilid cavefish. The nucleotide skews may have been the result of mutational and selective pressures during DNA replication and transcription(Perna and Kocher, 1995; Wei et al., 2010a, b). Thus,the unusual composition ofND6in the cavefish may be related to selective pressure from their extreme habitat. Nevertheless, further investigation is required to understand how nucleotide skews influence replication direction or gene orientation, and what selective pressures cause skews to arise.
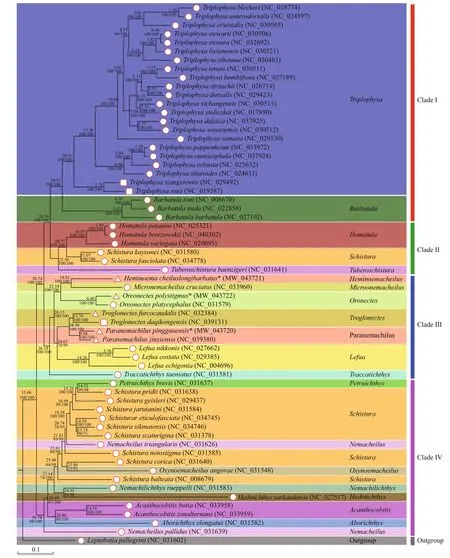
Fig.6 Phylogenetic tree and divergence time of Nemacheilidae based on sequences of 13 PCGs
The PCG Ka/Ks ratios were all <1 (Fig.4),suggesting that the PCGs evolved under purifying selection, during which harmful mutations with negative selection coeffi cients were naturally selected against (Yang and Bielawski, 2000). Under purifying selection, it is diffi cult for disadvantageous mutations to be established, and the long-term stability of biological structures is maintained. Thus, low Ka/Ks ratios forCOXI,COXIII, and CYTB indicate strong purifying selection and evolutionary conservation in these genes. Diff erences in Ka/Ks ratios can indicate diff erences in selection pressures and evolutionary mechanisms among genes (Lynch et al., 2006). The Ka/Ks ratios forATP8andND2were highest (Fig.4),suggesting thatATP8andND2evolved at a faster rate than the other mitochondrial PCGs in the nemacheilid cavefish. More functional genes are present in the H-strand of the mitogenome; for example,ATP8andND2both reside on the H-strand of the mitogenome,implying that selective pressures may have exerted stronger influence on the heavy chain than on the light chain in nemacheilid cavefish mtDNA.
Based on principal component analysis (PCA), the overall codon and amino acid usage patterns of the cavefish diff ered significantly from those of other fish(Fig.5e & f), further supporting the nucleotide composition and skew results. Mitochondria are the centres of energy metabolism in eukaryotic organisms and produce more than 95% of all cellular energy via oxidative phosphorylation (Xu et al., 2007). In cave habitats, the lack of autotrophic primary producers due to constant darkness can lead to limitations in food supply (Zhao and Zhang, 2009). The diff erences in nucleotide compositional biases between cavefish and other nemacheilid species suggest that mtDNA may have been subjected to evolutionary selection to improve energy use effi ciency in response to food limitations in cave environments; however, further investigations are required for this to be confirmed.
To explore the evolutionary history between cavefish and their surface-dwelling relatives, we constructed a phylogenetic tree based on the mitogenome PCGs of 61 nemacheilid species, including three species of stygobionts and five species of stygophiles, withLeptobotiapellegrinias the outgroup (Fig.6). Both maximum-likelihood (ML) and Bayesian inference(BI) approaches produced trees with similar topologies and branch lengths, indicating a well-resolved and highly supported phylogeny of Nemacheilidae. The data revealed thatHeminoemacheiluslongibarbatusandMicronemacheiluscruciatusclustered into one separate group, whileParanemachilusjinxiensisgrouped withParanemachiluspingguoensis. This corroborates the findings of Du et al. (2021) thatParanemachilusjinxiensis(formerlyYunnanilusjinxiensis) belongs to the genusParanemachilus. Our results also supportTroglonectesfurcocaudalisandTroglonectesdaqikongensis(formerlyOreonectesfurcocaudalisandOreonectesdaqikongensis,respectively) as species ofTroglonectes(Huang et al.,2020). Convergent evolution is a common phenomenon in extreme cave environments, and the present study challenges the results of previous morphological phylogenetic analyses (Wilkens and Strecker, 2003;Protas et al., 2006; Trontelj, 2019). Therefore,comprehensive taxonomic revisions of these species,using both morphological and genomic data, are needed.
The separation of cave and non-cave species(24.83 Ma-24.65 Ma-20.93 Ma-6.40 Ma) occurred concurrently with the early stages of the Miocene uplift of the southern Qinghai-Tibet Plateau (QTP),which was characterized by intensive intraplate tectonism, magmatism, and metallogeny (Li, 2010).Diff erentiation among cavefish occurred during the late stage of the Miocene uplift (13.76 Ma- 9.08 Ma-7.71 Ma), by which time the tectonism and magmatism that had defined the earlier stages were less prominent (Li, 2010). These splitting events coincided with the intraplate orogeny of the southern QTP, which mainly occurred 25 Ma- 7 Ma (Li, 2010).Throughout the Miocene uplift of the southern QTP,large-scale karst development occurred in the limestone regions (Shi et al., 1999). Uplift of the QTP significantly aff ected both the climate and habitat of the area (An et al., 2001; Xiao et al., 2005;Zhao and Zhang, 2009). Thus, the isolation of cave habitats, along with the unique environmental eff ects of the QTP uplift process, are considered key reasons for cavefish speciation (Zhao and Zhang, 2009; Yang et al., 2016). Our results provide further evidence that the speciation and diversification of nemacheilid cavefish are correlated with uplift of the QTP.
5 CONCLUSION
The complete mitogenomes of three cavefish(Order Cypriniformes, Family Nemacheilidae) were sequenced, annotated, and characterized. The mitogenome contained 37 genes (13 PCGs, 22 transfer RNAs (tRNAs), and two ribosomal RNAs (rRNAs))and a single control region. The Ka/Ks ratios indicate that the PCGs of the three cavefish evolved under purifying selection. The low Ka/Ks ratios forCOXI,COXIII, andCYTBindicate strong purifying selection and evolutionary conservation in these genes, whereas the high Ka/Ks ratios forATP8andND2indicate that these genes evolved at a faster rate than the other mitochondrial PCGs in nemacheilid cavefish. PCA revealed that the mitogenomes of the three nemacheilids exhibited nucleotide biases — within the PCGs, tRNAs, rRNAs, and full genomes. These results imply that the mtDNA underwent evolutionary selection to improve energy use effi ciency in response to food limitations in cave environments. Divergence time and evolutionary history analyses indicated that speciation and diversification of the cavefish coincided with the Miocene uplift of the southern QTP, when the cave habitats were undergoing significant change.This study has helped to elucidate the mitogenomes and phylogeny of fish in Nemacheilidae and provided insights into the evolutionary history of cavefish.
6 DATA AVAILABILITY STATEMENT
The complete mitochondrial DNA sequences were deposited in GenBank under accession numbers MW043720 (P.pingguoensis), MW043721 (H.longibarbatus), and MW043722 (O.polystigmus).The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
7 ACKNOWLEDGMENT
We are very grateful to Jiahu LAN from Du’an Fishery Technique Popularization Station for specimen collection and photographing on the three species.
 Journal of Oceanology and Limnology2022年4期
Journal of Oceanology and Limnology2022年4期
- Journal of Oceanology and Limnology的其它文章
- Validation and error analysis of wave-modified ocean surface currents in the northwestern Pacific Ocean*
- The energy conversion rates from eddies and mean flow into internal lee waves in the global ocean*
- Observation of physical oceanography at the Y3 seamount(Yap Arc) in winter 2014*
- Decadal variation and trend of the upper layer salinity in the South China Sea from 1960 to 2010*
- Observations of turbulent mixing and vertical diff usive salt flux in the Changjiang Diluted Water*
- Experimental research on oil film thickness and its microwave scattering during emulsification*