
Bacterial communities associated with hydromedusa Gonionemus vertens in diff erent regions in Chinese coastal waters*
2022-08-13 06:17WenjinHAOLeiWANGFanLITingtingSUNSaijunPENGYongxueLIJianminZHAOZhijunDONG
Wenjin HAO , Lei WANG , Fan LI , Tingting SUN , Saijun PENG , Yongxue LI ,Jianmin ZHAO ,4, Zhijun DONG ,4,**
1 School of Life Science, Nantong University, Nantong 226019, China
2 Muping Coastal Environment Research Station, Yantai Institute of Coastal Zone Research, Chinese Academy of Sciences, Yantai 264003, China
3 Shandong Key Laboratory of Marine Ecological Restoration, Shandong Marine Resource and Environment Research Institute,Yantai 264006, China
4 Center for Ocean Mega-Science, Chinese Academy of Sciences, Qingdao 266071, China
Abstract Bacteria communities in cnidarian jellyfish can be harmful to many important aquaculture species, as they can be key vectors of bacterial pathogens. However, our knowledge of bacterial communities associated with jellyfish in culture ponds and their potential roles in the regulation of aquaculture species remains unclear. In this study, sequencing based on the bacterial 16S rRNA gene was used to investigate the composition and variation of the bacterial communities associated with hydromedusa Gonionemus vertens in sea cucumber culture ponds and natural marine environment. The associated bacterial communities of G.vertens from the culture ponds in the Yellow Sea and Bohai Sea had significantly diff erent compositions,when compared with those from ambient seawater environment. Furthermore, bacterial communities associated with G. vertens had similar diversity and composition in culture ponds and natural marine environment in the Yellow Sea. There were 31 unique bacterial biomarkers identified in three locations.The major communities were highly abundant in Kiloniellales, Octadecabacter, Polynucleobacter, and Polaribacter, and are related to the environmental information processing. Pathogen candidates such as Vibrionales and Chlamydiales had notably low relative abundances (<1%). The venom of the jellyfish was considered responsible for damage to the aquaculture. This study provides important data to help assess the impact of cnidarians-associated bacterial communities on pond aquaculture and the influences on material cycling and energy flow in marine ecosystems.
Keyword: bacterial community; hydromedusa; the Yellow Sea; the Bohai Sea
1 INTRODUCTION
Jellyfish blooms are a growing concern from marine aquaculture industry, as they have been associated with an increasingly number of mortality events in Atlantic salmon farming, which has resulted in economic losses and fish welfare issues (Småge et al., 2018). In recent years, sudden fish mortalities in marine aquaculture were reported from Asia,Australia, and North and South America as results of episodic blooms of diff erent jellyfish species (Bosch-Belmar et al., 2017).
Gonionemusvertens(Hydrozoa, Limnomedusae)is a highly toxic cryptogenic clinging jellyfish which has outbreaks in coastal areas around the world and is considered an invasive species (Rodriguez et al.,2014; Gaynor et al., 2016; Govindarajan and Carman,2016; Govindarajan et al., 2017; Marchessaux et al.,2017).G.vertenswas introduced to the Mediterranean Sea, and has now invaded into the northern Europe(Bakker, 1980), the western North Atlantic(Govindarajan and Carman, 2016), and the western South Atlantic (Rodriguez et al., 2014), indicating that it can readily travelled and invade into new areas(Schuchert, 2016). Invasive species can have harmful impacts on local native taxa and disrupt normal ecosystem functioning (Gallardo et al., 2016;Govindarajan et al., 2017). In addition, they can also have direct negative impacts on human health (Ruiz et al., 2000; Pyšek and Richardson, 2010). The hydromedusae have a potent sting for causing severe pain and other symptoms in humans (Govindarajan and Carman, 2016; Marchessaux et al., 2017), and can be lethal to their predators (Carman et al., 2017).
A great number of jellyfish can clog fish cages by tidal movement, which would obstruct water exchange, resulting in oxygen depletion and jellyfish toxin buildup. Moreover, the stuck jellyfish could be torn off by ropes or wires of fish cages into toxic nematocyst-containing pieces that can pass through the mesh of the fish cages (Delannoy et al., 2011).Nematocysts could still be released from the pieces,which may be taken into or adhere to the fish, leading to severe lesions or fatal, especially when gills are infected (Ferguson et al., 2010). Jellyfish toxins are the most potent of all venoms, as they have cytotoxic,neurotoxic, cardiotoxic, hemolytic, dermatonecrotic,immunogenic, and inflammatory eff ects (Mariottini et al., 2008). If fish did not die directly from the toxins,they would succumb within a few hours due to respiratory failure or later from secondary bacterial infections to the body and the gills, caused by opportunistic bacteria such asFlavobacterimandVibrios(Ferguson et al., 2010).
Significant species-specific bacterial diff erences between native jellyfish (Aureliaaurita) and invasive gelatinous zooplankton (Blackfordiavirginicaand the comb jellyMnemiopsisleidyi) were confirmed, and the latter mainly consisted ofMycoplasmaandVibrio(Jaspers et al., 2020). The microbiome associated with jellyfishRhizostomapulmois identified in three major prokaryotic genera (Spiroplasma,Mycoplasma,andWolinella), which are considered bacterial vectors and potential hazards for marine animals and human health (Basso et al., 2019; Clinton et al., 2020).Cnidarian jellyfish may also be key carriers for bacterial pathogens asTenacibaculummaritimumthat was isolated fromPelagianoctilucawas implicated in cultured marine fish gill disease (Delannoy et al.,2011; Fringuelli et al., 2012).T.maritimumhas been reported to infect gills damaged by jellyfish venom(Delannoy et al., 2011). The pathogenesis and natural reservoir of this pathogen has not yet been clarified,and the associated lesions are characterized by necrosis in the mouth, head, fins, and gills (Delannoy et al., 2011). Pathogenesis of the lesions is thought to be the result of the proteolytic activity of extracellular toxins (Delannoy et al., 2011). The disease, which is recognized worldwide, is considered an important threat to aquaculture (Toranzo et al., 2005). However,the medusae were collected far away from sea-cages ofinfected salmon; since these jellyfish are small enough to pass the cages, it was impossible to determine the original source of the bacteria(Delannoy et al., 2011).
In the present study, we investigated the bacterial communities associated withG.vertensfrom two diff erent environments to determine if this hydromedusa species is a direct potential host of marine agriculture pathogen. We aimed to analyze the bacterial community compositions associated with hydromedusaG.vertensin both marine agriculture and natural environments using 16S rRNA sequencing,attempt to determine the diversity, composition, and potential functions of the bacterial communities, and to compare the associated bacterial communities from diff erent geographic regions. This study provides information regarding the associated bacterial communities of hydromedusaeG.vertensoutbreaks for the maintenance of environment and marine organisms.
2 MATERIAL AND METHOD
2.1 Animals collection and preparation
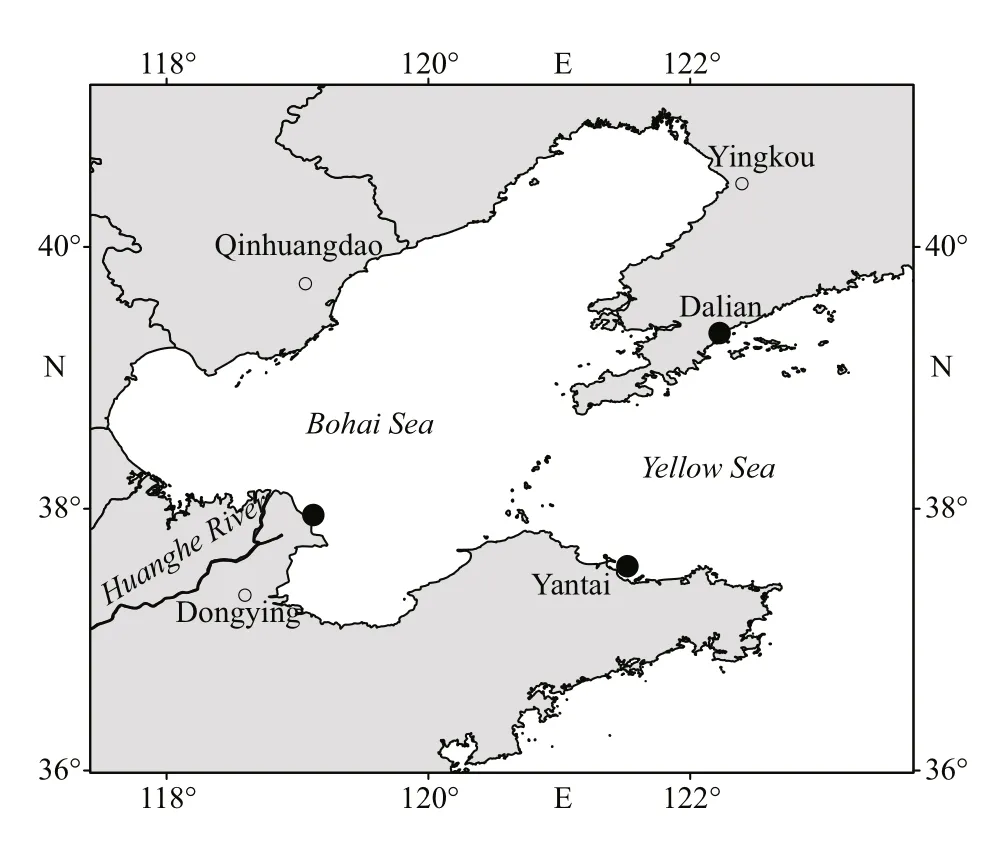
Fig.1 Sampling locations in the Chinese coastal waters
Gonionemusvertenswas sampled in two diff erent environments (Fig.1): sea cucumber culture areas in Dalian, Liaoning (coded GVDL; 39°34′21″N,122°22′47″E) and Dongying, Shandong (coded GVDY; 37°95′21″N, 119°12′05″E), and a natural marine environment in the Sishili Bay, Yantai,Shandong (coded GVYT; 37°56′09″N, 121°52′02″E),using a 500-μm dip net towed onboard a research vessel with long-handled bucket. In each station, 10 intactG.vertensspecimens were taken and immediately placed into sterile seawater (pre-filtered by 0.2-μm pore size filter and autoclaved) baths for 3 h to clear their guts and remove loosely associated microorganisms. Each specimen was rinsed three times with sterile seawater before further analysis,stored at -20 °C, and freeze-dried before DNA extraction. Seawater samples (500 mL) collected from each location at the same time as seawater sample (coded as SWDL, SWDY, and SWYT,respectively to the three areas ofG.vertenssampling)were pre-filtered by 3-μm pore size filters (TCTP, 47 mm, Millipore, Germany) and then sequentially filtrated through 0.2-μm pore size filter (Millipore Corporation, Bedford, MA) with three parallels to obtain free-living bacterial community from water column for comparison to the jellyfish-associated bacteria (Daniels and Breitbart, 2012). The bacterial biomass from the filters was stored at -80 °C until further processing.
2.2 DNA extraction and PCR amplification
Total genomic DNA from the bacterial community associated with each individualG.vertenswere extracted from freeze-dried tissues using cetyltrimethyl-ammonium bromide (CTAB) as per Hao et al. (2015) with slight modifications. The filter membranes of each seawater sample were applied to extract the total DNA from the seawater samples using a PowerWater DNA Isolation Kit (MOBIO,USA) according to the manufacturer’s instructions.The hypervariable regions V4-V5 of the bacteria 16S rRNA gene were amplified with specific primers:515F (5′-GTG CCA GCM GCC GCG GTA A-3′) and 907R (5′-CCG TCA ATT CCT TTG AGT TT-3′). All PCR reactions were carried out using the Phusion High-Fidelity PCR Master Mix (New England Biolabs) with 0.2 μmol/L of each primer and 10-ng template DNA. Thermal cycling consisted ofinitial denaturation at 98 °C for 1 min, followed by 30 cycles of denaturation at 98 °C for 10 s, annealing at 50 °C for 30 s, and elongation at 72 °C for 30 s, followed by 72 °C for 5 min (Gong et al., 2019). All PCRs were performed in triplicate, and no-template controls were included in all steps of the process. The PCR products were detected by electrophoresis in a 2% (w/v)agarose gel. The PCR amplicons of each sample that produced bright bands were mixed in equal density ratios and purified with GeneJETTM Gel Extraction Kit (Thermo Scientific). The amplicon libraries were generated using Ion Plus Fragment Library Kit 48 rxns (Thermo Scientific) and sequenced using the IonS5TMXL platform at Novogene Bioinformatics Technology Co., Ltd. (Beijing, China).
2.3 Bioinformatics and Statistical Analyses
The analysis was conducted by QIIME2docs along with customized program scripts (https://docs.qiime2.org/2019.1/). Briefly, raw FASTQ files were demultiplexed using the QIIME2 demux plugin based on the unique barcodes. Demultiplexed sequences from each sample were quality filtered and trimmed,de-noised, merged, and then the chimeric sequences were identified and removed using the QIIME2 dada2 plugin to obtain the feature table of the amplicon sequence variant (ASV) (Gong et al., 2019). The QIIME2 feature-classifier plugin was then used to align the ASV sequences to a pre-trained SILVA database (trimmed to the V4-V5 region bound by the 515F/907R primer pair) to generate the taxonomy table (Bokulich et al., 2018). Any contaminating mitochondrial and chloroplast sequences were filtered using the QIIME2 feature-table plugin. Linear discriminant analysis (LDA) eff ect sizes (LEfSe),accounts for both bioconsistency and significance,were used to identify the significant diff erences in the bacterial taxa between the sea cucumber breeding area (GVDL and GVDY) and the natural marine environment (GVYT). Diversity metrics were calculated using the core-diversity plugin within QIIME2. Feature level alpha diversity indices, such as observed operational taxonomic units (OTUs),Chao1 richness estimator, Shannon diversity index,and faith_pd were calculated to estimate the microbial diversity within an individual sample (Gong et al.,2019). Beta diversity distance, including Bray Curtis and Weighted UniFrac were measured to investigate the variation of the bacterial communities structure among all samples, and visualized using principal coordinate analysis (PCoA). The Venn diagram was applied to screen out common OTU among diff erentsample groups. Major bacterial communities were filtered with the relative abundant higher than 1%from these common OTU. Functional diversity of the bacterial communities associated with jellyfishG.vertenswas predicted using PICRUSt (Langille et al.2013). OTUs were assigned with QIIME’s command“pick_closed_otus” with 97% similarity in Greengenes13.5 database. Then, the predicted functions were blasted to the Kyoto Encyclopedia of Genes and Genomes (KEGG) database at group levels 1 and 2, and statistical diff erences among groups were analyzed using test package of R language.ANOVA+Duncan and Dunn tests were conducted to analyze whether there was significant diff erence in the prediction function of microbial community among groups, unless specified above, the default parameters were used in the analysis.
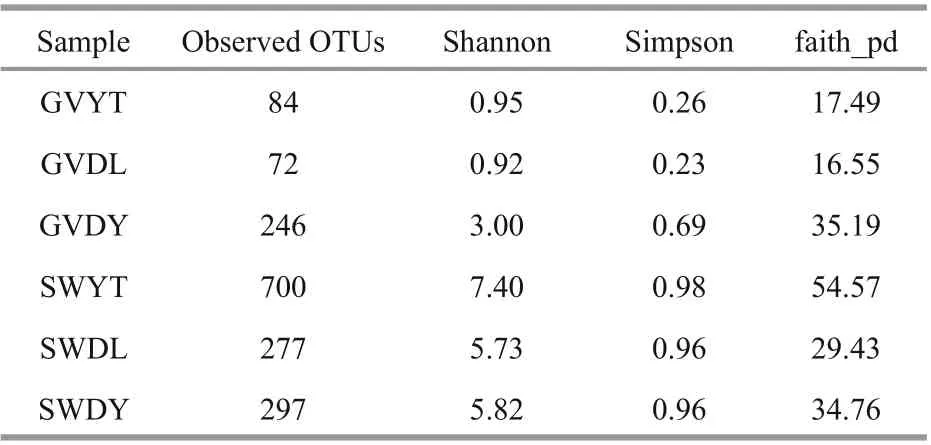
Table 1 The observed OTUs and diversity indices (Shannon,Simpson, and faith_pd) retrieved from sequencing data for diff erent geographic regions
3 RESULT
3.1 Sequencing and classification
PCR products for the V4-V5 regions were sequenced using the single-end method with the IonS5TMXL platform, and this generated a total of 3 060 720 raw tags representing 39 samples, with individual reads ranging from 53 229 to 93 790.After qualification and removal of the chimeras from the raw tags, a total of 2 920 866 validated sequence reads, and 74 894 for each sample on average(ranging 51 493-89 956), and an average of 370 bp,were obtained. The eff ective tags of all samples were grouped into 58 278 OTUs based on 99% identities.Through normalized processing, a total of 4 039 OTUs were retained for all further downstream analyses. The trend of the rarefaction curves suggested that there was suffi cient sampling of the bacterial communities, as it showed that each sample was diff erent (Supplementary Fig.S1 & Table S1).This indicated that the bacterial diversity was well revealed by the high sequence number per sample,which was enough to acquire the majority of the 16S rRNA gene sequences.
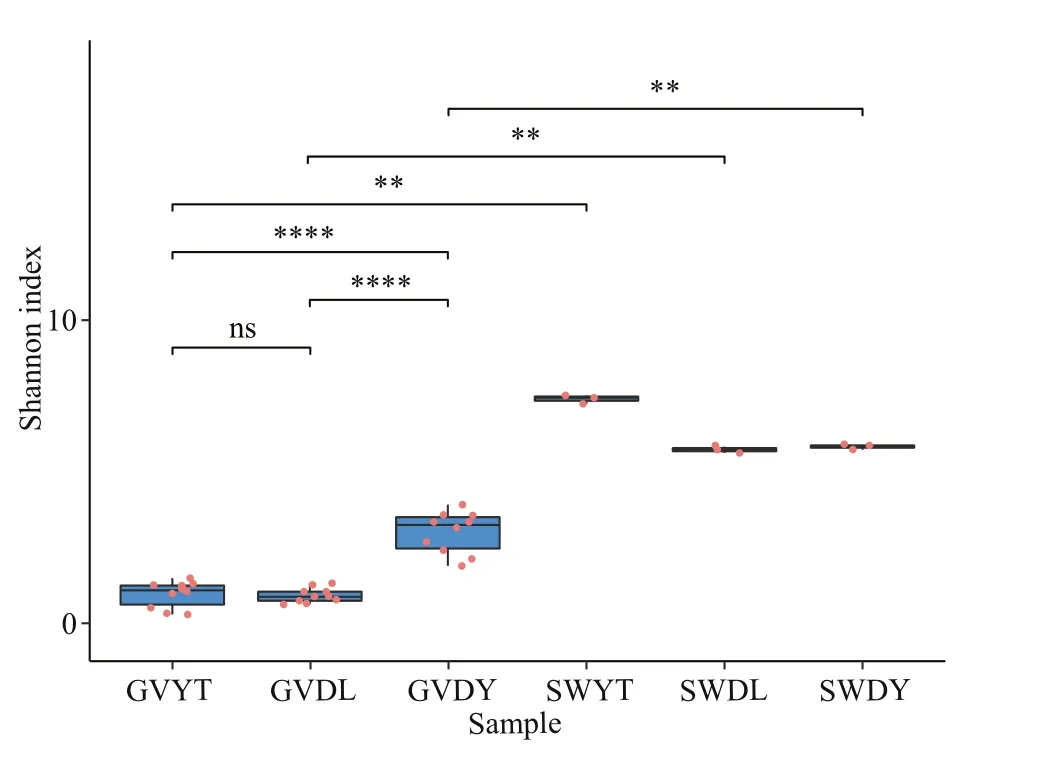
Fig.2 Alpha diversity of the bacterial communities associated with G. vertens and the environment seawater, represented by the Shannon index accompanied by multiple comparisons based on the Wilcox test
3.2 Analysis of alpha diversity
The alpha diversity of all samples is displayed in Table 1. The lowest OTUs were 72 in the GVDL samples and the highest were 246 in the GVDY samples among jellyfish samples. Moreover, the Shannon index of the GVDY samples was the highest(3.00). Compared with the natural environment samples, the SWYT sample had the highest value(7.40). We found that SWYT also had the highest Simpson indices (0.98) and faith-pd indices (54.57).According to the multiple comparisons based on Wilcox test, we found that the Shannon index of the GVDY was significantly diff erent from that of GVDL and GVYT (P<0.001). In addition, the alpha diversity of the jellyfish samples at three locations (GVDL,GVDY, and GVYT) were significantly diff erent from those of the seawater samples (SWDL, SWDY, and SWYT), respectively (Fig.2).
3.3 Beta diversity analysis
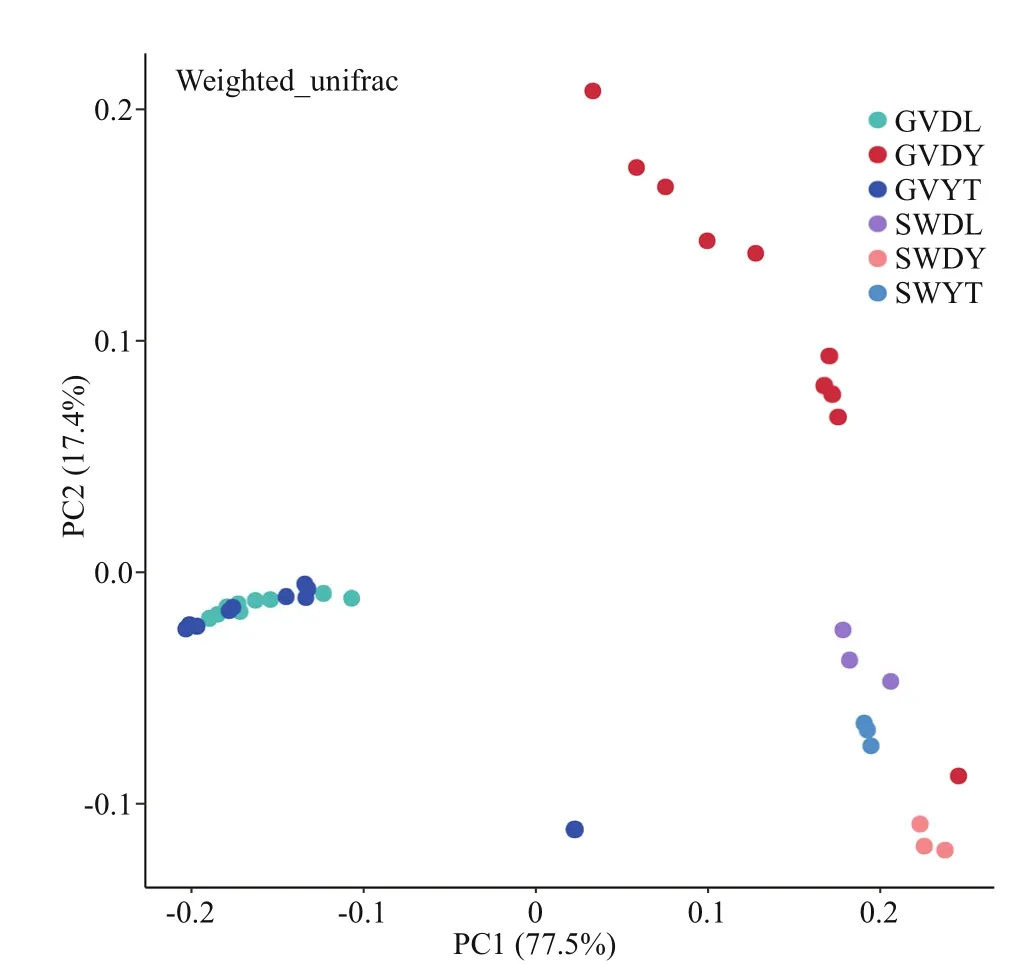
Fig.3 Principal coordinate analysis (PCoA) presenting the bacterial communities associated with G. vertens and seawater from diff erent locations based on weighted unifrac metrics
Principal coordinate analysis (PCoA) was performed to visualize and compare the structure of the bacterial communities among diff erent samples.The PCoA results based on weighted unifrac distances demonstrated that the jellyfish samples (GVDL,GVDY, and GVYT) and seawater samples (SWDL,SWDY, and SWYT) tended to separate according to PC1 (77.5%) and PC2 (17.4%) (Fig.3). The bacterial communities associated with GVDL clustered with those from GVYT, but they were all separated from the corresponding seawater samples. The PERMANOVA main test indicated significant diff erences among the bacterial communities associated withG.vertensamong the three diff erent regions (P=0.001; Table 2), where pair-wise comparisons indicated that the bacterial community associated with the GVDY was significantly diff erent from the communities associated with GVDL and GVYT (Supplementary Table S2;P=0.001).
3.4 Bacterial community composition in G. vertens
The relative abundances of the identified taxa from the phylum to order were reported as stacked bar plots for each analyzed sample (Fig.4a-c). Only taxa with a relative abundance equal to or higher than 1% were plotted, and the rest was collapsed into “other”.
Proteobacteria dominated the observed sequences at the phylum level for jellyfish samples, representing 95.31%, 95.08%, and 90.95% of the total number of species in GVDL, GVDY, and GVYT, respectively,followed by Bacteroidetes accounting for 4.35%,1.18%, and 4.01% of GVDL, GVDY, and GVYT,respectively. Furthermore, Bacteroidetes were found the predominant phylum in SWYT (52.25%), SWDL(46.06%), and SWDY (44.53%), followed by proteobacteria in the SWDL (45.89%), SWYT(37.51%), and SWDY (29.27%). Meanwhile,Actinobacteria levels were also high in the SWDY,SWDL, and SWYT samples, accounting for 20.62%,6.34%, and 4.89%, respectively, followed by Cyanobacteria for 3.83% of the SWDY (Fig.4a).
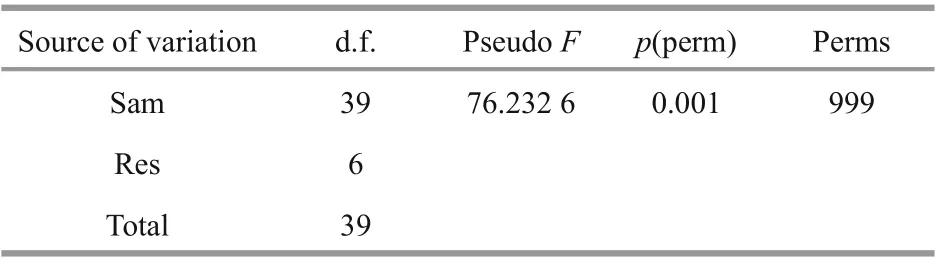
Table 2 PERMANOVA main tests of the bacterial community structures associated with G. vertens and seawater from diff erent locations based on weighted unifrac dissimilarities
Within Proteobacteria, Alphaproteobacteria were the most dominant group (91.42%) at the class level in the GVDL sample, followed by GVYT (88.63%) and GVDY (59.33%). Flavobacteriia made up a small portion of the GVDL (4.3%), GVYT (3.9%), and GVDY (0.55%). In addition, the level of Betaproteobacteria was high in GVDY samples(22.17%), followed by Epsilonproteobacteria (9.95%)and Gammaproteobacteria (3.24%) (Fig.4b).Concerning the natural environment seawater samples,Flavobacteriia occupied a large part of the relative abundance of SWYT (46.01%), SWDL (42.5%), and SWDY (31.55%) (Fig.4b). Alphaproteobacteria levels were also high in SWDL (40.82%), SWYT (24.67%),and SWDY (19.82%), while the Actinobacteria made up 20.28% of SWDY (Fig.4b).
Kiloniellales was the most abundant group in the GVDL (87.59%) and GVYT (83.98%) (Fig.4c). In GVDY, Rhodobacterales and Burkholderiales were found the most dominant group accounting for 48.75% and 21.98%, respectively, followed by Campylobacterales (9.95%). In addition,Flavobacteriales and Rhodobacterales were dominant in the samples of seawater samples of SWDL (42.50%and 32.32%), SWDY (31.55% and 13.11%), and SWYT (46.01% and 20.81%). Actinomycetales accounted for 20.28% in SWDY, while Rickettsiales appeared in all SWDL, SWDY, and SWYT samples,accounting for 6.58%, 4.35%, and 1.78%, respectively(Fig.4c).
3.5 LEfSe analysis revealed specific bacterial biomarkers
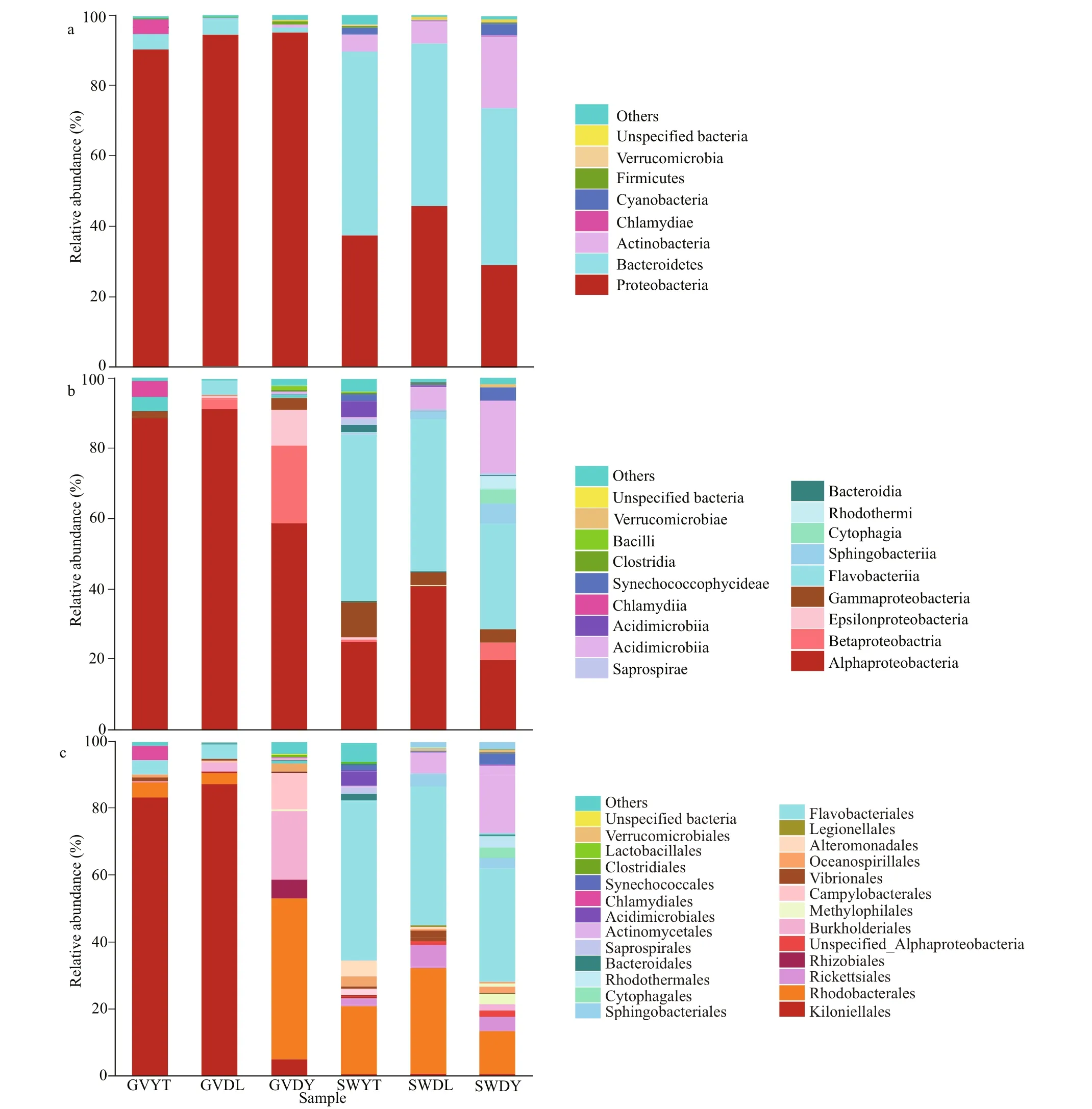
Fig.4 Stacked bar-plots of relative abundances of the bacterial communities compositions associated with jellyfish G. vertens and the environment seawater at three diff erent locations at phylum (a), class (b), and order (c) levels
Based on the above analyses of the diversity,similarity, and compositions of the bacterial communities associated withG.vertensamong three locations, linear discriminant analysis (LDA) and the eff ect sizes (LEfSe) were applied to reveal the bacterial bioindicators that were significantly associated withG.vertensin the two diff erent environments at the genus level. We identified 31 unique bacterial biomarkers (Fig.5) in the bacterial communities associated withG.vertensamong the three locations using the LEfSe analysis (LDA score>4 andP<0.05). Five bacterial taxa, including Rhodobacteraceae and Oxalobacteraceae, and its two members (i.e.,PseudoruegeriaandPolynucleobacter)were detected as biomarkers in GVDL. Thirteen diff erent taxa were identified as discriminant biomarkers of GVDY. Six and three out of the nine biomarkers identified in GVDY belong to Alphaproteobacteria (e.g.,OctadecabacterandLabrenzia) and Epsilonproteobacteria (e.g.,Arcobacter), respectively. In addition,Pseudonocardiabelong to Actinobacteria, and the biomarkers in the GVYT included Streptococcaceae, andPolaribacterbelonging to Bacteroidetes, andVibrioandRoseobacterbelonging to Proteobacteria.
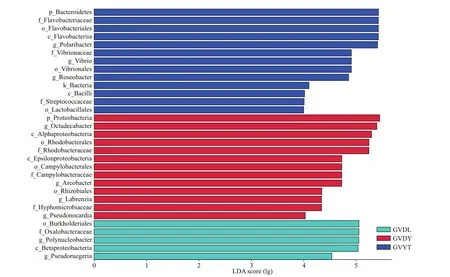
Fig.5 LDA eff ect size of the LEfSe analysis to identify potential bacterial bioindicators present at the genus level in G. vertens in diff erent environments (LDA score >4 and P <0.05)
3.6 Major bacterial communities in G. vertens
We further analyzed the core microbiome fractions from the three locations. The shared and unique OTUs among diff erent samples were shown in the Venn graphs (Supplementary Fig.S2). There were 2 405 OTUs in total, which accounted for 59.54% of the total 16S rRNA gene reads ofG.vertens. The OTU number drastically dropped to 55 when a restriction of each OTU (>1%) was observed in all locations of this study, and the 55 shared OTUs were regarded as the core OTUs. However, when only those with a relative abundance higher than the threshold value of 0.5% were considered, the number of core OTUs dropped rapidly to 14. According to our results, the major community was highly abundant with Kiloniellales (64.83% and 1.23%) andOctadecabacter(13.00% and 0.92%, respectively) (Fig.6). The predominant colonizers were assigned toPolynucleobacter(7.12%),Polaribacter(2.83%),Rhodobacteraceae (1.60%),Arcobacter(1.76% and 1.24%), and Chlamydiales (1.6%) (Fig.6).
3.7 Predictive analysis of bacterial function
Using the Kyoto Encyclopedia of Genes and Genomes ortholog pathways (Langille et al., 2013),the KEGG functions of the identified bacteria were determined to be significantly (P<0.05) among three locations (Fig.7). The results showed four potential functional categories including environmental information processing, metabolism, genetic information processing, and unclassified, respectively.There were 25 pathways identified that were involved in metabolism, and purine metabolism, peptidases,oxidative phosphorylation, butanoate metabolism,and amino sugar and nucleotide sugar metabolism were diff erent among the three locations. In genetic information processing, DNA repair and recombination proteins and ribosome biogenesis were diff erent between the GVDL, GVYT, and GVDY.
4 DISCUSSION
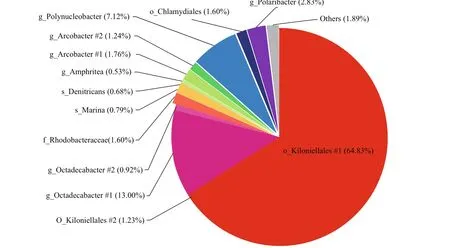
Fig.6 The major species bacterial communities (>0.5% relative abundance) of G. vertens among the three locations higher than 0.5% in relative abundance
Previous studies characterizing the bacteria associated with marine invertebrates focused largely on benthic organisms such as corals and sponges.They all contain microbiota that are distinct from the ambient water, and these bacteria play critical roles in host ecology (Cleary et al., 2016). In the present study, we characterized the bacterial communities associated with hydromedusaG.vertensthat were collected from three diff erent locations: two sea cucumber mariculture environments (Dongying and Dalian) and a natural marine environment (Sishili Bay, Yantai). The bacterial communities associated withG.vertenshad a lower diversity as indicated in the 16S rRNA gene sequencing than that of the environmental seawater (Fig.2 & Table 1). The bacterial communities associated with jellyfish from Dongying had the highest number of observed OTUs(246) and the highest Shannon index (3.00) when compared to the other two locations (Table 1). Cortés-Lara et al. (2015) and Viver et al. (2017) reported a reduced diversity of the microbiota associated with the gastric cavity of the jellyfishCotylorhizatuberculata. As clarified by Tinta et al. (2019) and Daley et al. (2016), the lower diversity in the microbial communities was associated with the invasive hydrozoanNemopsisbacheiand the cosmopolitan scyphozoanAureliaauritahosted taxon-specific bacterial groups. The bacterial community associated with ctenophore contained less bacterial OTUs and lower diversity communities than that in water column (Daniels and Breitbart, 2012). The low diversity of the jellyfish-associated bacterial communities diff ered from the trend previously observed in corals and sponges, in which bacterial diversity was typically higher than that of the surrounding water column (Rohwer et al., 2002;Taylor et al., 2007; Lee et al., 2011). Although these gelatinous animals are primarily composed of water,it has previously been reported that their associated bacterial communities are strikingly diff erent from the surrounding water column (Daniels and Breitbart,2012; Hao et al., 2015; Weiland-Bräuer et al., 2015;Daley et al., 2016; Lee et al., 2018; Tinta et al., 2019).In the present study, the bacterioplankton communities both from the natural environment (Sishili Bay) and the aquiculture area (the holothurian mariculture areas in Dongying and Dalian) had high diversity(observed OTUs, Shannon, and Simpson index) and were significantly distinct from the jellyfishassociations (P=0.001). The bacterial communities obtained from the natural environment (Sishili Bay)had higher observed OTUs (700) and Shannon’s indexes than those in the sea cucumber mariculture areas in Dongying (297) and Dalian (277). The ANOSIM and PERMANOVAP-values indicate no overlap among the bacterial communities from jellyfish and from surrounding water.
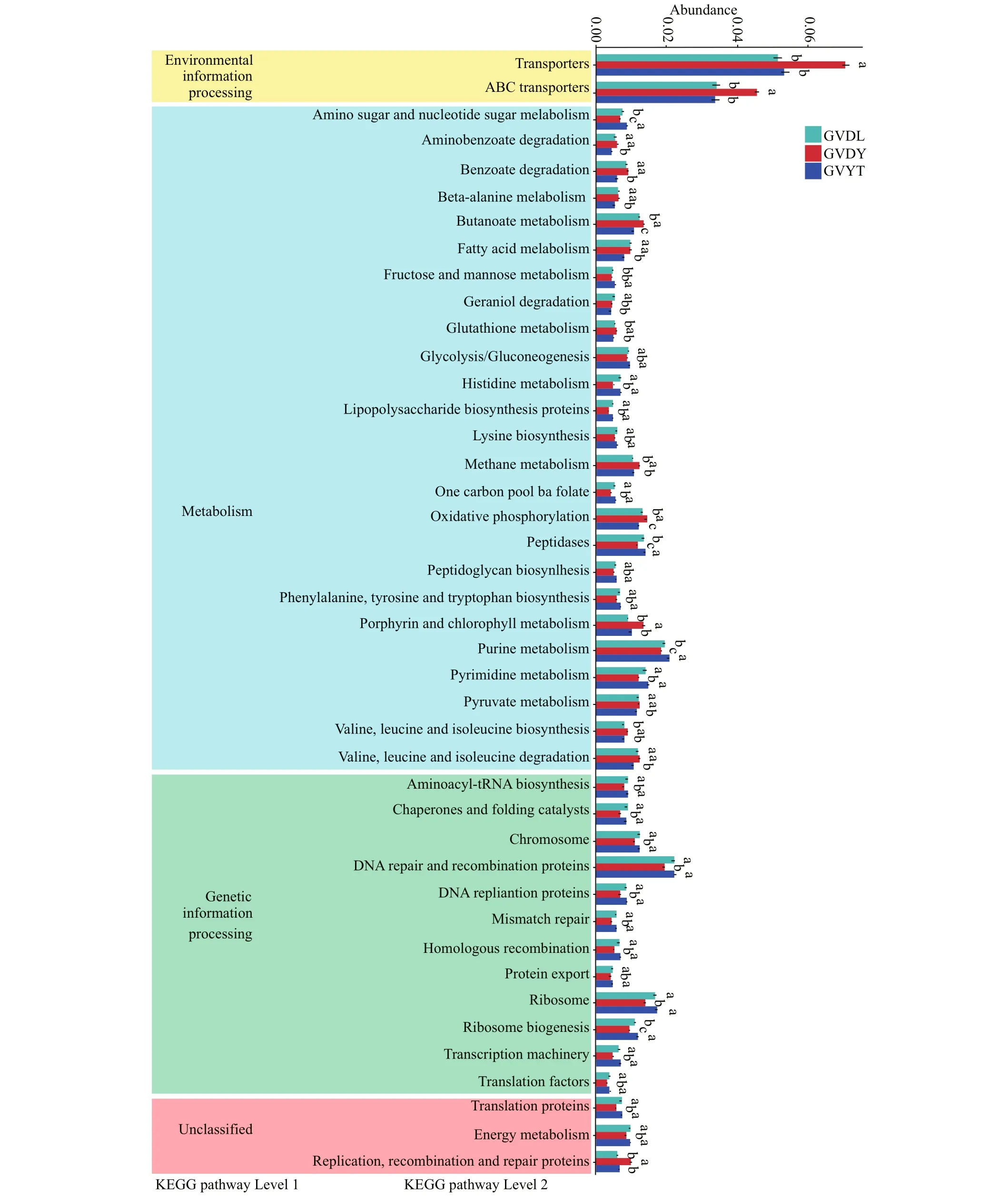
Fig.7 The relative abundance of predicted KEGG functional pathway profiles based on PICRUSt among three locations
The bacterial communities associated withG.vertensfrom the holothurian mariculture area(GVDL) and natural environment (GVYT), collected in the Yellow Sea, mainly consisted of Alphaproteobactereia. The communities from the holothurian mariculture area (GVDY) from the estuary area in the Bohai Sea were composed of Alphaproteobacteria, Betaproteobacteria, and Epsilonproteobacteria. The Huanghe River estuary and coastal habitats of the Bohai Sea are some of the most productive marine ecosystems on earth and suff er greatly from upstream and coastal anthropogenic pollution. Such area are extremely dynamic with steep physicochemical gradients due to the variability of the freshwater input and geomorphology. Due to the diff erences in geographic environments and ecological conditions among diff erent sea areas, the microbial community structures and characteristics are diversified (Webster et al., 2015). Many abiotic factors shape the microbial distribution patterns, such as salinity (Webster et al., 2015), temperature (Lindh et al., 2013), pH (Wang et al., 2015a), depth (Walsh et al., 2016), nutrient status (Fodelianakis et al., 2014),spatial factors (Martiny et al., 2006), biological resources, and the marine environment (Wei et al.,2016). Diff erentiated gradients control the bacterial diversity, abundance, and biogeographic distribution,in which microbes are crucial for maintaining functional and structural balances in estuarine and open-sea ecosystems through active biogeochemical cycling and complex food webs (Webster et al., 2015;Wei et al., 2016). It was demonstrated in this study that spatial separation and environmental condition decided the bacterial communities and they were distinctly diff erent in Dongying and Dalian in the Bohai Sea and the Yellow Sea, respectively, whereas in the jellyfish that were collected from the sea cucumber culture ponds in diff erent sea areas, they had similar structures in Dalian and Yantai both in the Yellow Sea area. It has been reported that the intestinal flora of the sea cucumberApostichopusjaponicusis influenced by specific geographical environments of the Yellow Sea and Bohai Sea (Ye et al., 2018).However, Hao et al. (2015) reported that a common member of the bacterial community of ctenophora that was closely related to Marinomonas species,occurred in diff erent regions, e.g., Tampa Bay, the Gulf of Mexico, USA, the Gullmar fjords of Sweden,and German Bight in Germany. There has been much controversy concerning vertical and horizontal transmission of these associated microbial community(Martiny et al., 2006). Mixed strategies combine the best horizontal and vertical transmission modes and play an important role in the evolution and ecology of host-symbiont relationships (Russell et al., 2017;Bernasconi et al., 2019b). Environmental changes in physical disturbances, seasonality, and climate aff ect microbial symbionts composition directly or via the physiological responses of the host, which, in turn,lead to changes in the conditions of boundary layers microhabitat (Wahl et al., 2012). For instance, the off spring of host can directly benefit the symbionts through vertical transmission. Unfortunately, this may have adverse eff ects if the transmitted association is not optimal in diff erent environment conditions from those of their parents. Meanwhile, horizontal transmission may help the host to take over diverse associations well fitted to the specific environment(Byler et al., 2013). It has been shown that there is a mechanism for microbial community isolation and diff erentiation in the marine environment. Ocean currents, geographical distance, seafloor landform,life history, diff erential genetic drift, and sharp changes in temperature and salinity could aff ect gene flow, resulting in isolation and genetic diff erentiation of diff erent microbial species (Webster et al., 2010).Therefore, diff erent jellyfish species who can use both strategies (horizontal transmission and vertical transmission) are more likely to have physiological and evolutionary advantages than those strictly relying on one or another (Bernasconi et al. 2019a).
Gammaproteobacteria, Bacteroidetes, and Alphaproteobacteria were found dominated in the bacterial sequences of hydromedusaNemopsisbachei(Daley et al., 2016). Concerning the bacterial communities associated withG.vertensin Dongying,the most abundant alphaproteobacterial OTUs inhabiting jellyfish were assigned to orders Rhodobacterales (48.75%), Burkholderiales(21.98%), Campylobacterales (9.95%), Rhizobiales(5%), and Oceanospirllales (2%). In contrast, the bacterial communities associated withG.vertensfrom Yantai and Dalian were very similar, and dominated by Kiloniellales. Moreover, only 5% of Kiloniellales were present in jelly-associated community in Dongying station. Cleary et al. (2013)found that three OTUs assigned to Kiloniellales took more than 63% of the total bacterial community of sponge in marine lakes in the Kakaban and Maratua islands in Indonesia. Afterwards, they compared the bacterial communities in two jellyfish species (the‘golden’ jellyfishMastigiascf.papua, and the box jellyfishTripedaliacf.cystophora) from marine lakes in the Berau region in the northeastern Borneo,Indonesia, and found that they were dominated by OTUs assigned to Gammaproteobacteria, Mollicutes,Spirochaetes, and Alphaproteobacteria. The most abundant alphaproteobacterial OTUs were assigned to orders Kiloniellales and Rhodobacterales. OTUs assigned to the order Kiloniellales had their greatest relative abundance in Tripidelia hosts (Cleary et al.,2016). Kiloniellales were rare or absent in the seawater samples from the natural environment in the present study.Kiloniellawas first reported by Wiese et al. (2009) as a novel alphaproteobacterium, and was isolated from marine macroalga. The genusKiloniellarepresents the type of the new family Kiloniellaceae fam. nov. and order Kiloniellales ord.nov. (Wiese et al., 2009). Phylogenetically, the genusKiloniellais related to the type strains of three Thalassospira species (88.9%-89.2%), which have been described previously in cnidaria-associated bacterial studies (Cortés-Lara et al., 2015; Hao et al.,2015). Kiloniellales is mesophilic and chemoheterotrophic aerobe with the potential for denitrification, and it displays a typical marine growth response (Wiese et al., 2009). All members of the Kiloniellales were found in quite diff erent marine habitats and were obtained from sponges (Jang et al.,2015), shrimp (Wang et al., 2015b), spider crab(Gerpe et al., 2017), and algae (Si et al., 2017). Wiese et al. (2009) provided genome insights into several metabolic properties, such as carbon and sulfur metabolism, and indicated the potential for denitrification and biosynthesis of the secondary metabolites ofKiloniella.
In the present study, only a fewVibrioand Chlamydiae were detected as biomarkers in Yantai station whereG.vertenswere collected from a natural marine environment. We did not detect any pathogen sequences in the bacterial communities associated withG.vertensin the sea cucumber culture pond in the Dongying and Dalian stations. Hydrozoan siphonophoreMuggiaeaatlanticaand microscopic hydromedusaePhialellaquadrataandSolmariscoronahad crucial impact on fish mortality in Irish and Scottish fish farms (Baxter et al., 2012; Bosch-Belmar et al., 2017). In parallel, it has been hypothesized that ctenophores and jellyfish can serve as vectors for fish pathogens, and recent findings have revealed that bacteria associated with hydromedusa served as the source of secondary infections in farmed salmon (Ferguson et al., 2010; Delannoy et al., 2011).Schuett and Doepke (2010) reported that the tentacles of HydrozoaTubulariaindivisiawere associated with potential endobiotic pathogenic bacteria, such asCobetiamarina,Colwelliaaestuarii,Endozoiciminaselysicola,Vibrioaestuarianus,Bacillussubtilis, andIlyobacterpsychrophilus(Schuett and Doepke, 2010).The most abundant sequences were affi liated with the genusTenacibaculum, in which 20 species have been isolated from marine environments. Five species were from water and four from sediment samples, others species, such asT.crassostreaeandT.adriaticumwere associated with healthy bryozoans, sea anemones, oysters, sponges, and green algae(Ferguson et al., 2010; Viver et al., 2017). In particular,T.maritimumis a known fish pathogen that has been detected in HydrozoaM.atlantica(Fringuelli et al.,2012) andP.quadrata(Ferguson et al., 2010), and is implicated in farmed fish gill disease. However, we did not detect any sequence similarity with this species in our study. The impact ofG.vertenson mariculture, mainly its toxic stings that can cause neuroparalysis and anaphylactic shock, and the eff ects of the pathogenic bacteria (e.g. the cultivation and specific phylogentic analysis of known pathogens)need to be studied in future studies. Carman et al.(2017) reported that the mortality of spider crabs increased withGonionemusconsumption, and 100%of the spider crabs died within 24 h after consuming jellyfish with toxic eff ects, suggesting thatGonionemussp. medusae can feed on hard-bodied organisms such as copepods and cladocerans as well(Govindarajan et al., 2019). It is notorious for causing severe stings in humans and is considered invasive or cryptogenic elsewhere. In general, we speculate that the toxicity of hydromedusaG.vertenson sea cucumbers was probably caused by the nematocysts more than pathogenic bacterial infections.
5 CONCLUSION
Jellyfish-associated bacterial communities were less diverse than both ambient seawater environments(sea cucumber mariculture area and natural environment), and their compositions were distinct from bacterioplankton communities in seawater.Rhodobacterales, Burkholderiales, Campylobacterales,Rhizobiales, and Oceanospirllales were abundant in the bacterial communities associated withG.vertensin Dongying. In contrast, bacterial communities from Yantai and Dalian were dominated by Kiloniellales,which is related to denitrification and biosynthesis of secondary metabolites. The associated bacterial communities had pronounced diff erences in diff erent geographical environments; and spatial separation and environmental eff ects were two critical factors. In addition, pathogen sequencing was not detected in the holothurian mariculture system of the symbiosis of jellyfishG.vertens. HydromedusaG.vertensmight had a great toxicity impact on sea cucumbers than that of the pathogen bacterial infections. Further studies need to reveal more details of pathogenic bacterial inG.vertensand other jellyfish species concerned.
6 DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon request.
 Journal of Oceanology and Limnology2022年4期
Journal of Oceanology and Limnology2022年4期
- Journal of Oceanology and Limnology的其它文章
- Validation and error analysis of wave-modified ocean surface currents in the northwestern Pacific Ocean*
- The energy conversion rates from eddies and mean flow into internal lee waves in the global ocean*
- Observation of physical oceanography at the Y3 seamount(Yap Arc) in winter 2014*
- Decadal variation and trend of the upper layer salinity in the South China Sea from 1960 to 2010*
- Observations of turbulent mixing and vertical diff usive salt flux in the Changjiang Diluted Water*
- Experimental research on oil film thickness and its microwave scattering during emulsification*