
Seasonal co-occurrence patterns of bacteria and eukaryotic phytoplankton and the ecological response in urban aquatic ecosystem*
2022-08-13 06:17JingYANGXiongjieZHANGJunpingQiLIUFangruNANXudongLIUShulianXIEJiaFENG
Jing YANG ,XiongjieZHANG , Junping LÜ, Qi LIU, Fangru NAN, Xudong LIU,ShulianXIE,JiaFENG
School of Life Science, Shanxi University, Taiyuan 030006, China
Abstract Microorganisms play a key role in aquatic ecosystems. Recent studies show that keystone taxa in microbial community could change the community structure and function. However, most previous studies focus on abundant taxa but neglected low abundant ones. To clarify the seasonal variation of bacterial and microalgal communities and understand their synergistic adaptation to diff erent environmental factors, we studied the bacterial and eukaryotic phytoplankton communities in Fenhe River that runs through Taiyuan City, central China, and their seasonal co-occurrence patterns using 16S and 18S rDNA sequencing. Results indicate that positive interaction of eukaryotic phytoplankton network was more active than negative one except winter, indicating that the cooperation (symbiotic phenomenon in which phytoplankton are interdependent and mutually beneficial) among them could improve the adaption of microbial community to the local environmental changes and maintain the stability of microbial network. The main genera that identified as keystone taxa in bacterial network were Salinivibrio and Sphingopyxis of Proteobacteria and they could respond to the variation of nitrite and make use ofit, while those that identified as keystone taxa in eukaryotic phytoplankton network were Pseudoschroederia and Nannochloris, and they were more susceptible to nitrate and phosphate. Mychonastes and Cryptomonas were closely related to water temperature. However, the loss of the co-occurrence by environmental factor changes aff ected the stability of network structure. This study provided a reference for analyzing relationship between bacteria and eukaryotic phytoplankton and revealing potential importance of keystone taxa in similar ecological domains in carbon, nitrogen, and phosphorus dynamics.
Keyword: seasonal co-occurrence; bacteria and eukaryotic phytoplankton communities; keystone taxa;ecological eff ect; urban aquatic ecosystem
1 INTRODUCTION
Urban water bodies are important to human life.Unfortunately, they are suff ering serious pollution in recent decades due to anthropogenic activities, posing a threat to the health of aquatic ecosystems, and having changed the physical and chemical environment and microbial community composition(Gücker et al., 2011; Drury et al., 2013). Previous researches have demonstrated that eutrophication by nitrate, phosphate, ammonium, and other organic matter pollutant (permanganate and suspended solids,etc.) had adverse eff ects on the functions of ecological community and aquatic ecosystem (Toyama et al.,2020; Zhang et al., 2020). As an important part of an aquatic ecosystem, microbial community plays an irreplaceable role in participating in biogeochemical processes and nutrient cycling.
In a natural ecosystem, an individual organism does not exist alone, but coexists with each other to form a network of ecological interactions (Mikhailov et al., 2019). These complex associations ease the influence of biodiversity on ecosystem functions(Duff y et al., 2007; Du et al., 2020). Microorganisms(such as bacteria and microbial eukaryotes) are important contributors to biodiversity as ecological role-players involved in primary production, food web, and biogeochemical cycle (Xue et al., 2018).Community structure is not only aff ected by interactions between species, but also by environmental fluctuations and regional conditions(Jones et al., 2013; Capo et al., 2017). In addition, the patterns of co-occurrence of bacterial and eukaryotic phytoplankton communities revealed based on nextgeneration sequencing can predict whether the ecological interactions among environmental variables and various species in aquatic ecosystems are positive or negative (Paver et al., 2013; Bunse et al., 2016), which is important for understanding the structure of microbial communities and provide insights into potential interaction networks (Ma et al.,2016).
Network analysis can determine the keystone species that are necessary to maintain community stability among many species. Vick-Majors et al.(2014) and Martín González et al. (2010) demonstrated that the nodes with higher betweenness centrality values are particularly important in maintaining the connectivity of ecological network and identify them as keystone species. A keystone species is a species that plays a critical and unique role in local natural environment although its abundance may not be high(Paine, 1969). Keystone species can aff ect the entire microbial community through a series of pathways.For example, they can aff ect the community structure and function through adjusting some intermediate groups and eff ect groups. Besides, keystone species are also susceptible to dynamic environments, the disappearance or change of keystone species may cause disturbance to mature communities (Steele et al., 2011).
At present, there have been numerous reports on the temporal and spatial changes and co-occurrence dynamics of microbial communities (Hu et al., 2017;Jiao et al., 2020), but most of them focused on abundant taxa. A microbial community is composed of a few abundant taxa and many rare taxa. Abundant taxa are contributive to the organic matter flux and biomass yield (Pedrós-Alió, 2012), whereas certain rare taxa may include more metabolically active microorganisms, which might be considered as keystone species that regulate the function of aquatic environment (Wang et al., 2020). In addition, rare taxa contribute to the stability and persistence of the community, becoming the “seed bank” of microbial and may become dominant under favorable conditions(Shade and Gilbert, 2015). At present, many studies have realize the importance of rare taxa in microbial ecology. For example, Jousset et al. (2017) reported that in terms of taxonomy and functional diversity,rare taxa might increase functional redundancy and enhance the ability of communities against environmental interference, which is critical to the nutrient cycles. Mallon et al. (2015) pointed out that some indigenous rare taxa in the environment could control the invasion of foreign microorganisms (such as pathogens), and indicated that rare microorganisms play an irreplaceable role in the ecosystem. However,due to the limitation in method, the rare biosphere has been largely unexplored. Recently, rapid development of next-generation sequencing and deep sequencing promoted the research on rare biosphere communities,and the ecological importance of rare sub-community attracted widespread attention (Wu et al., 2017).
One of the main purposes of current microbial ecological research is to understand the formation and maintenance mechanism of microbial diversity(including abundance and composition). On this issue, the most mainstream view is that the microbial community structure is influenced by both deterministic processes based on niche theory(including species interactions, environmental selection, etc.) and stochastic processes based on neutral theory (including diff usion limitation, drift,etc.) (Jiao et al., 2020). The neutral theory predicts that community structure and composition are relevant to the geographic distance between samples,which is due to the dispersal restrictions and many species have equal functions in ability to utilize a niche. According to the niche theory, changes in species community composition are related to variations in environmental parameters (Dumbrell et al., 2010), because species with unique characteristics could enable themselves to develop unique habitats for surviving.
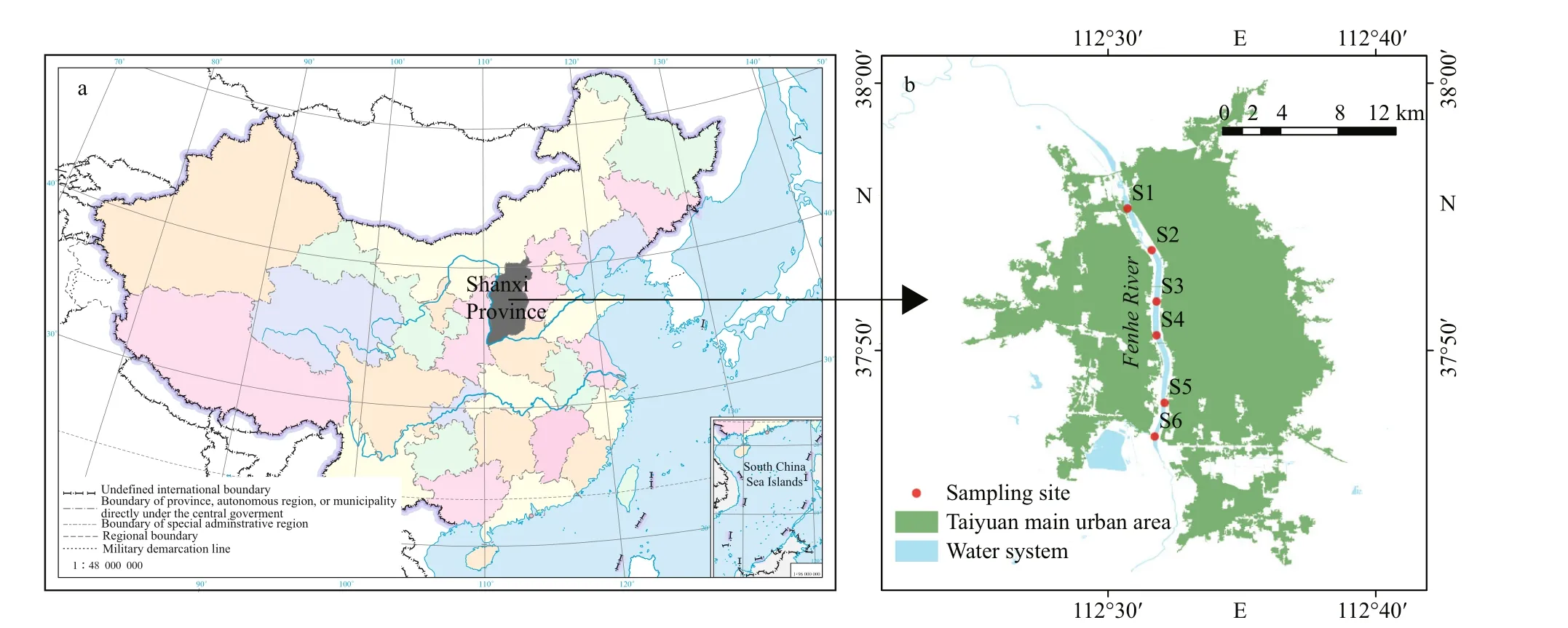
Fig.1 Location of the study area and the sampling sites along the Fenhe River in Taiyuan City
The interrelationship networks are complex and can contain the inherent mechanism of microbial interaction in environmental disturbance transmission(Hunt and Ward, 2015). Therefore, identifying the interactions between bacteria and microeukaryotes and the combination of complex ecological communities is critical for the management of biodiversity, especially in the areas of strong human interference and fragile ecosystem. Although many studies are conducted on the interaction between abundant taxa (Ger et al., 2016), few are on how rare taxa aff ect the community structure of microorganisms in aquatic ecosystem. In addition, knowledge on cooccurrence patterns of rare taxa in urban water bodies remains poor. As the capital of Shanxi Province,Taiyuan is an important coal industrial base in China.Due to the excessive development and consumption of coal resources in the recent decades, the ecological environment in urban Taiyuan has been seriously polluted. At present, the microbial network in the Taiyuan section of the Fenhe River has not been systematically analyzed. Therefore, 16S and 18S rDNA gene sequencing was used to detect the seasonal pattern of bacterial and eukaryotic phytoplankton communities in the study area, and network analysis was used to identify keystone species. Moreover, the species were divided into abundant taxa and rare taxa,from which relationship between keystone taxa and environmental variables are discussed. This research will help understand the interactions and ecological eff ects of microorganisms and improve our ability to predict the response of microorganisms to environmental changes.
2 MATERIAL AND METHOD
2.1 Sampling and environmental parameter
Fenhe River is the first tributary of the Huanghe(Yellow) River with 39 471 km2in area and 710 km in total length. The study area is located in Taiyuan(37°27′N-38°25′N, 111°30′E-113°09′E) (Fig.1). The regional climate is characterized by a semi-humid continental monsoon climate belonging to a semienclosed water body. The mean annual precipitation is 504.8 mm. Besides, it is also the main coalproducing area, industrial area, and urban residential area of Shanxi Province.
For this study, 144 samples in 6 sampling stations were collected in 2019 on March 22, April 18, and May 14 for spring; on June 10, July 17, and August 12 for summer; on September 16, October 11, and November 19 for autumn; and on January 8, December 16 for winter. Three replicates were set for each sample. A 2-L sampler was used to col1ect water samples at the depth of 0.5 m, and then 1 000 mL of water volume was filtered through 0.22-μm polycarbonate filters (Milipore, USA). The >0.22-μm fraction was used for DNA extraction, and the corresponding water samples were used to determine the physical and chemical parameters. Water temperature and pH were measured by a multiparameter water quality meter (Hydrolab DS5, Hach Company, USA). Nitrate (NO3ˉ), nitrite (NO2ˉ), and phosphate (PO43ˉ) were measured as per Wang et al.(2008). The concentration of dissolved organic carbon(DOC) was determined using total organic carbon analyzer (Shimadzu Corporation, Kyoto, Japan). The water and wastewater monitoring analysis method(Zhu et al., 2019) was used to measure chlorophyll-a(Chl-a) concentrations. The significance (One-way ANOVA) of each parameter was calculated by SPSS 26.0.
2.2 DNA extraction, PCR amplification, and products purification
Genomic DNA was extracted from environmental samples using E.Z.N.ATMMag-Bind DNA Kit(OMEGA) as per manufacturer’s instructions.Agarose gel was used to detect the purity and concentration of DNA extracts, and then the qualified samples were selected for subsequent analysis. The V3-V4 regions of 16S rDNA gene was amplified using primers 341F (5′-CCTACGGGNGGCWGCAG-3′) and 805R (5′-GACTACHVGGGTATCTAATCC-3′) (Herlemann et al., 2011); the V4 region of 18S rDNA gene was amplified using primers V4F(5′-GGCAAGTCTGGTGCCAG-3′) and V4R(5′-ACGGTATCTRATCRTCTTCG-3′) (Sun et al.,2014). The PCR amplification cycle was: the first round of amplification was 94 °C for 3 min, followed by 5 cycles at 94 °C for 30 s, 45 °C for 20 s, 65 °C for 30 s, then followed by 20 cycles at 94 °C for 20 s,55 °C for 20 s, 72 °C for 30 s, and final extension at 72 °C for 5 min. The second round of amplification was 95 °C for 3 min, followed by 5 cycles at 94 °C for 20 s, 55 °C for 20 s, 72 °C for 30 s, and a final extension at 72 °C for 5 min. Two-round PCR reactions were carried out in 30-μL solution including 15-μL Phusion High-Fidelity PCR Master Mix,0.2 μmol/L of forward and reverse primers, and 10-ng genomic DNA or PCR products of first-round amplification. Samples were then sequenced on the Illumina MiSeq sequencing platform in the Sangon Biotech Co., Ltd. (Shanghai, China), and paired-end reads were generated by sequencing the forward and reverse strands of each target DNA fragment. The rarefaction curve of each sample in this study tended to be flat, and the coverage rate reached 99.9%,indicating that the depth of sequencing reads was large enough.
2.3 Processing sequence data
The QIIME 2 software package (version 2019.1)was used to process raw sequence data (Bolyen et al.,2019). The DADA2 inference algorithm can perform the primer-free reads to correct sequencing errors and create amplicon sequence variants (ASVs) for microbial communities (Callahan et al., 2016).Therefore, DADA2 was used to perform quality control, denoising, filtering, merging, and removing chimeras on these sequences to generate ASVs. ASVs are more sensitive, specific, and repeatable, and using ASVs can better distinguish ecological patterns than the OTU method (Callahan et al., 2017). All raw sequence data in this study were submitted to the NCBI Sequence Read Archive (SRA) database under the accession number SRP273360.
2.4 Statistical analysis
In this study, the diversity index Simpson and Shannon index for α-diversity analysis was calculated using Mothur software. The significance (ANOVA variance) analysis followed by Tukey’s HSD post hoc tests of all indexes and environmental parameters was conducted by SPSS 26.0. Analysis of similarity(ANOSIM) was used to perform diff erences in microbial communities between groups by “vegan”package for R (version 4.0.3). The classification of rare and abundant species relies on the cutofflevel of relative abundance, ASVs with relative abundance greater than 1% are defined as abundant taxa, less than 0.01% is defined as rare taxa (Zhang et al., 2018).Abundant taxa including always abundant taxa (AAT)and conditionally abundant taxa (CAT). Rare taxa including always rare taxa (ART) and conditionally rare taxa (CRT).
The co-occurrence network of each season was established through Molecular Ecological Network Analysis (MENA) based on the relative abundance of ASVs (Deng et al., 2012), by which the Pearson Correlation Coeffi cients (PCC) were computed for each ASV pair, and then the statistical significance of PCC values was calculated in permutation test. The similarity threshold of microbial communities was determined according to the method of random matrix theory. The Benjamini-Hochberg method was used to standardize the correlation coeffi cients to correct the significanceP-value obtained from the original hypothesis test, and finally the correctedPvalue was used to keep the relevant ASV ofP<0.05 to construct the correlation network. Moreover, we used the Network Analyzer tool to obtain network topology parameters such as the characteristic path length, the number of connections, the number of nodes, the aggregation coeffi cient, the network density, and the average connectivity of the network.Edges were set between pairs of ASVs, for which the PCC was significant. Then, the networks were visualized with Gephi (Version 0.9.2) (Heymann,2014) and Cytoscape (version 3.2) (Shannon et al.,2003). Meanwhile, to compare the diff erence between the molecular ecological network and the corresponding random network, the topological properties based on Erdós-Réyni random networkwere calculated using R (Version 4.0.3) (Lieberman et al., 2005).
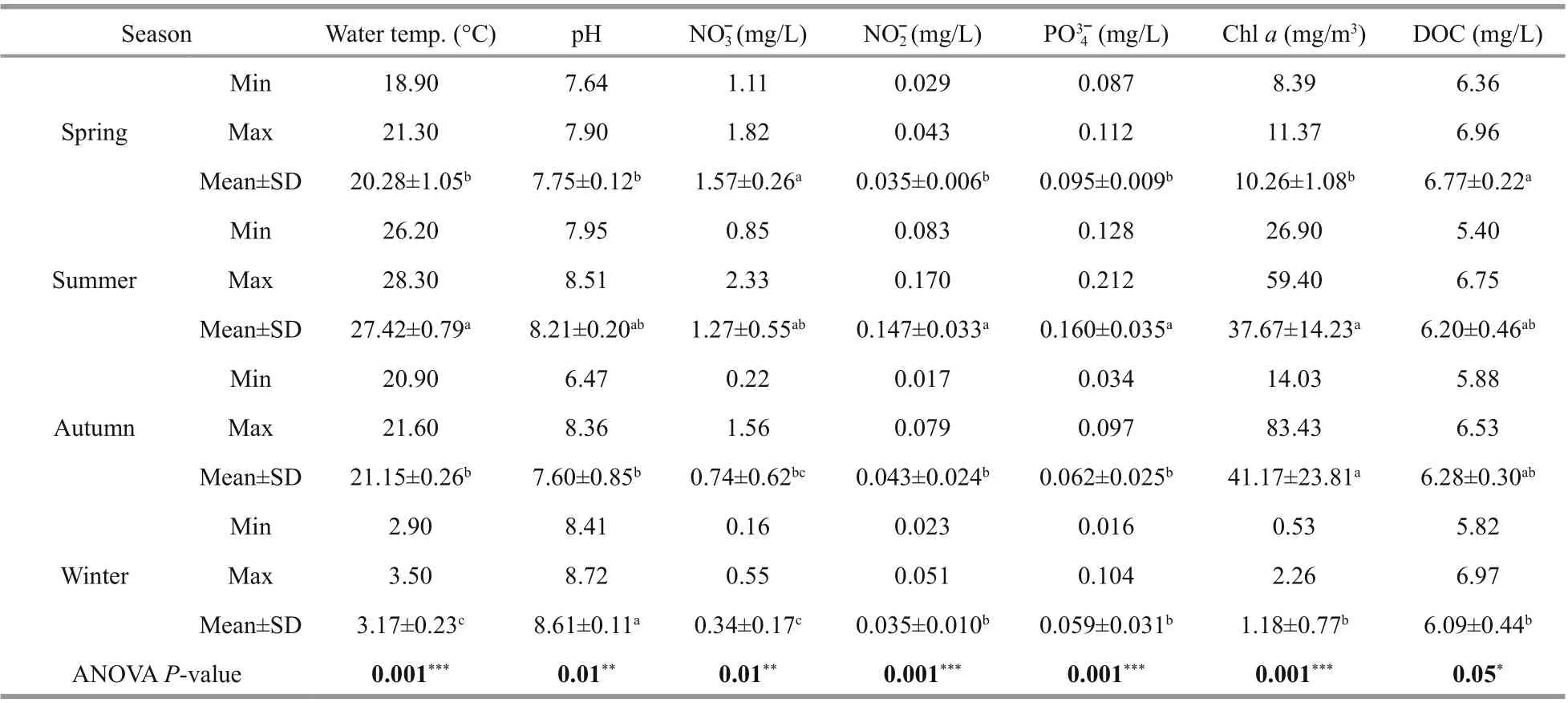
Table 1 Description statistics of main physical and chemical variables in the Fenhe River
In addiiton, we used the greedy modularity optimization method to characterize the network modularity of each network established in each season(Shi et al., 2016). Modules greater than 0.4 were used as the threshold of the detection module. To determine the topological properties of each node in the network,the nodes were divided into four categories through their among-module connectivity (Pi) and withinmodule connectivity (Zi): network hubs are nodes that are highly connected to other nodes in the entire network (Zi>2.5;Pi>0.6), module hubs are nodes that are highly connected to other nodes in the network modules (Zi>2.5;Pi<0.6), connectors are nodes that connect diff erent modules (Zi<2.5;Pi>0.6), and Peripherals are nodes that have less connectivity with other nodes (Zi<2.5;Pi<0.6) (Deng et al., 2012).
Ordination analysis was performed by Canoco 5.0. Detrended correspondence analysis (DCA) was used to determine whether unimodal or linear ordination methods should be applied. Results show that the relationship between the bacteria and environmental variables was only applicable to the unimodal model (canonical correspondence analysis,CCA), and the relationship between the eukaryotic phytoplankton and the environmental variables applied to both redundancy analysis (RDA) and CCA. The Monte Carlo permutation tests (499 permutations) were applied to identify the environmental variables with significant eff ects(Sarker et al., 2013).
3 RESULT
3.1 Physical, chemical, and biotic characteristics of water quality
The characteristics of physical, chemical, and biotic are shown in Table 1. The lowest values of nitrate, nitrite, phosphate, Chla, and DOC were detected in winter. Nitrate concentration in spring was generally higher than those in other seasons. Nitrite and phosphate concentrations in summer ranged 0.083-0.170 mg/L, 0.128-0.212 mg/L, respectively,which were also higher than those in other seasons.Water temperature was highest in summer, with an average of 27.4 °C, and post hoc HSD tests indicated there was no significant diff erence between spring and autumn. The pH value in winter was higher than in other seasons. The level of Chlain autumn was much greater than that in spring and winter (P<0.01).The maximum average DOC concentration was detected in spring. One-way analysis of variance showed that all parameters were significantly diff erent(P<0.05), and the DOC concentration in spring was significantly higher than that in winter (P<0.05).
3.2 Sequence quality and α-diversity indexes
The rarefaction curve obtained by measurement of the primers in this study tended to be flat, indicating that the amount of sequencing data was suitable. In addition, the coverage rate can also reflect whether the sequencing results represent the true situation of the sample. The actual test results of the primers allthat a significant diff erence existed across four seasons. Similarly, at the ASV level of bacteria,ANOSIM gave the results ofR2=0.89,P<0.01(between seasons) andR2=0.11,P>0.05 (between sites). The α-diversity indexes of bacteria and phytoplankton are shown in Supplementary Table S2.ANOVA test showed significant diff erences in diversity between seasons, indicating that when environmental factors change in season, and the species composition and abundance also change significantly. From the perspective of bacteria or phytoplankton, post hoc HSD tests indicated that the biodiversity in spring was significantly higher than that in autumn (P<0.05). The positive correlation between nitrate and DOC and Shannon index indicated that the structure and composition of the microbial community in this study mainly respond to the fluctuation of carbon and nitrogen.
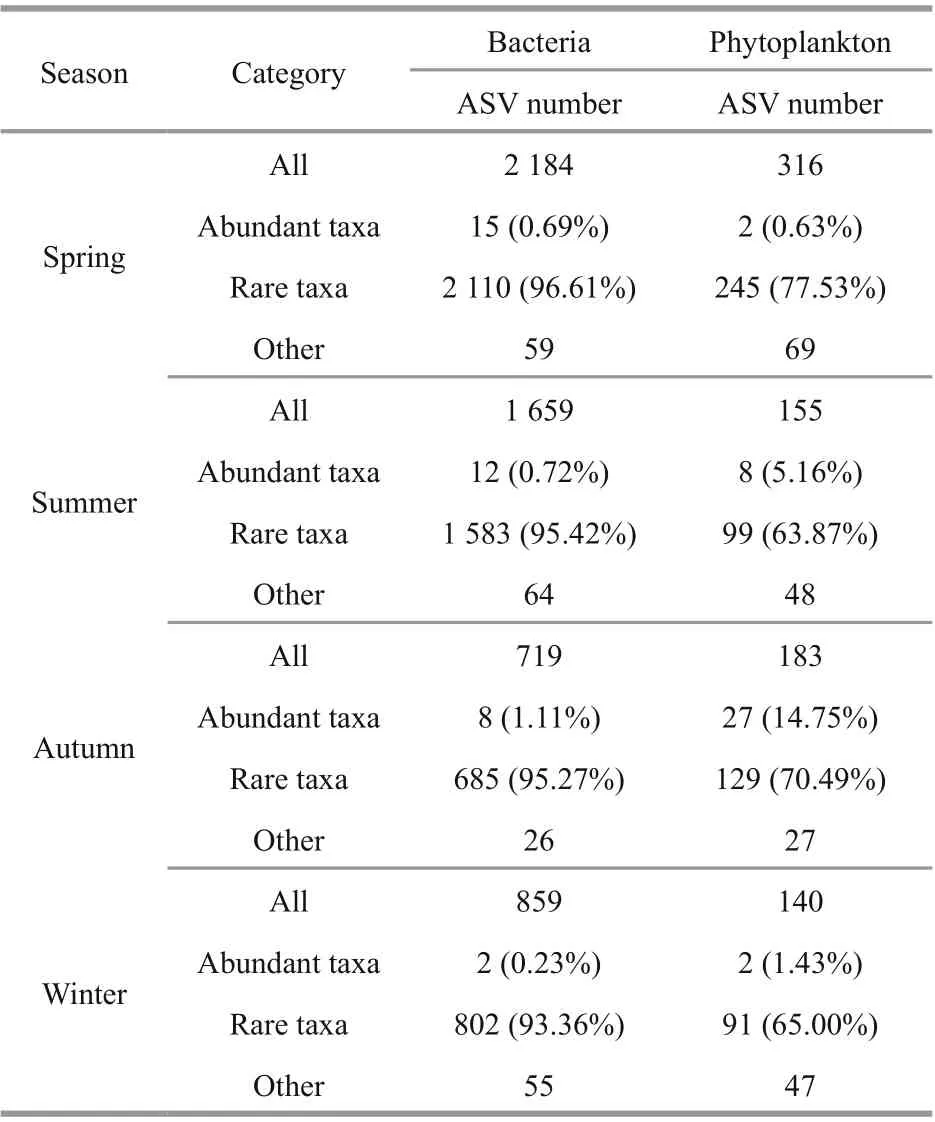
Table 2 General description of all, abundant, and rare ASVs datasets
3.3 Seasonal variation of bacterial and eukaryotic phytoplankton community
reached more than 99%, which reflected the real situation of the community composition in the tested sample. In this study, information of abundant taxa and rare taxa is shown in Table 2. Singletons were deleted before further analysis. The results show that the abundant taxa for bacteria accounted for only 0.23%-1.11% of the total sequences, while the number of rare taxa was more than 93% of total ASV number. Abundant taxa for phytoplankton accounted for 0.63%-14.75%, while rare taxa accounted for more than 63.87%. It can be seen that the number of rare taxa was much greater than those of abundant taxa.
The average number of high-quality sequences was the highest in autumn, 53 756 16S rDNA sequences and 23 690 18S rDNA sequences. However,the number of ASVs was higher in spring, with 327 for bacteria and 128 for eukaryotic phytoplankton(Supplementary Table S1). Sequences of 16S rDNA had an average length of 420 bp. Sequences of 18S rDNA had a mean length of 400 bp. At the ASV level of phytoplankton communities, ANOSIM was used to test the temporal and spatial similarity, and gave the results ofR2=0.92,P<0.01 (between seasons) andR2=0.04,P>0.07 (between sites), which demonstrated
The bacterial community composition at the genus level is shown in Fig.2. Our results demonstrate that Proteobacteria was the most abundant bacterial phylum in all seasons. Besides, Bacteroidetes were also the dominant phylum in spring. Dominant bacteria in summer samples included Actinobacteria and Firmicutes, and autumn samples included Bacteroidetes and cyanobacteria. The relative abundances of Planctomycetes and Verrucomicrobia in winter were higher than those in other seasons. At higher taxonomic resolution, the most enriched genus based on relative abundance included Proteobacteria(Acinetobacter,Brevundimonas,Comamonas,Citrobacter), Bacteroidetes (Cloacibacterium) in spring, of whichAcinetobacterandComamonaswere the abundant taxa,BrevundimonasandCitrobacterthe rare taxa; Proteobacteria (Acinetobacter,Comamonas,Pseudomonas, andSphingomonas),Bacteroidetes (Chryseobacterium), Firmicutes(Exiguobacterium) in summer, of whichAcinetobacter,Pseudomonas, andSphingomonaswere the abundant taxa,Comamonasthe rare taxa.The relative abundance ofAcinetobacterwas the highest in all samples in autumn, accounting for 80%-90%, onlyAcinetobacter,Delftia, andKlebsiellawere the abundant taxa, all other bacteria were the rare taxa. The dominant genus in winter diff ered greatly from that in other seasons, mainly including Proteobacteria (Janthinobacterium,Pseudomonas,andUndibacterium), Bacteroidetes (Flavobacterium),
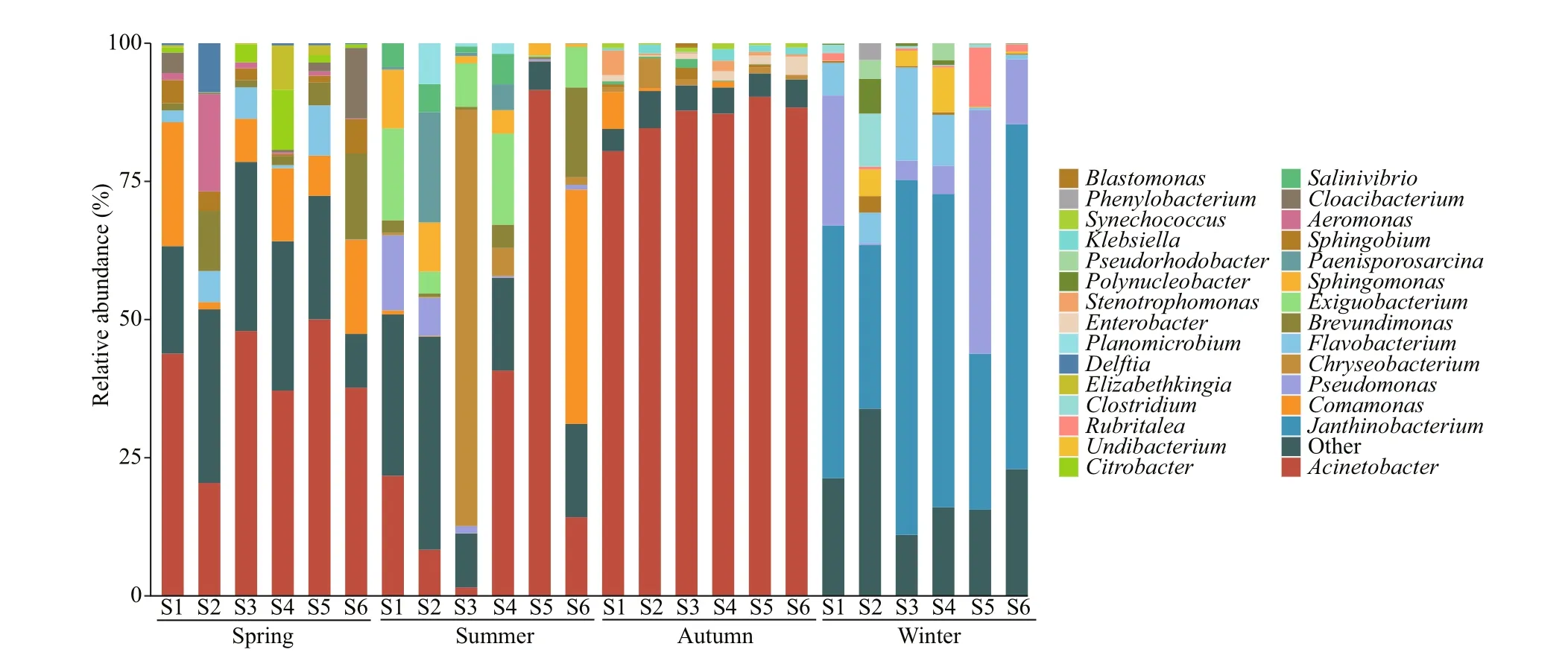
Fig.2 The relative abundance (%) of 16S rRNA gene reads allocated to a genus per sampling site
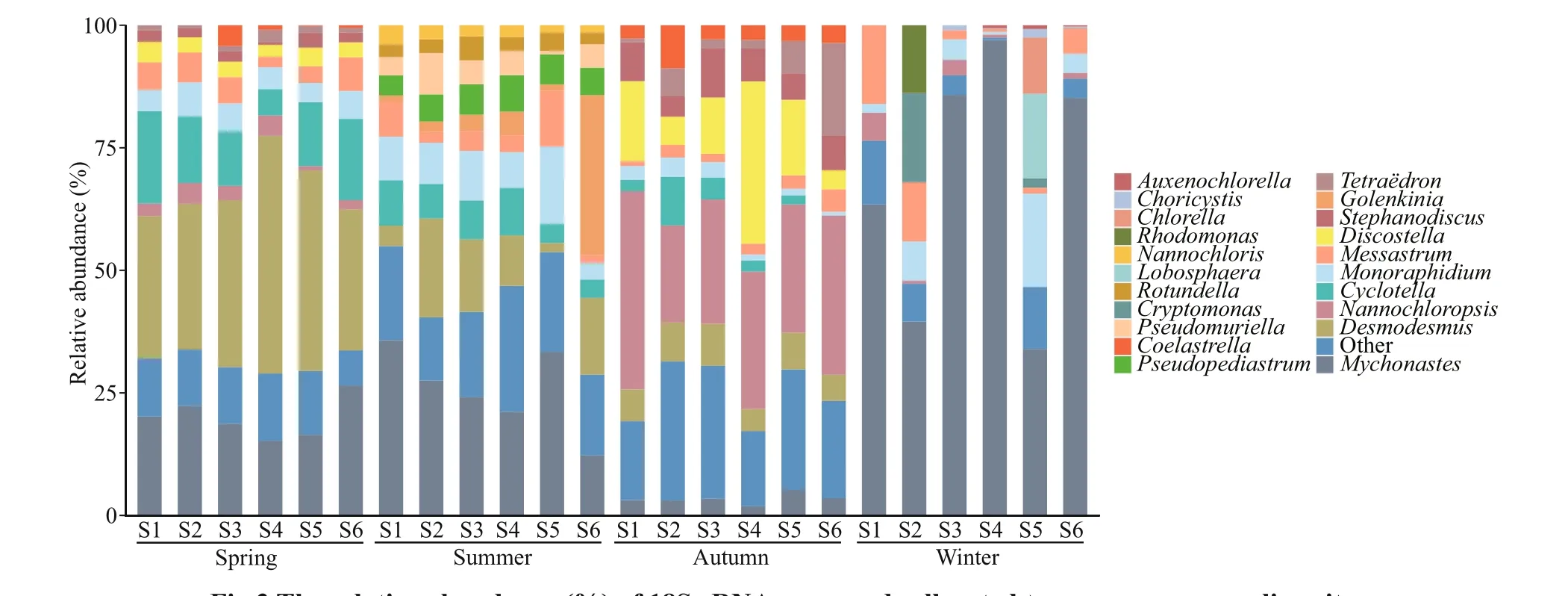
Fig.3 The relative abundance (%) of 18S rRNA gene reads allocated to a genus per sampling site
Firmicutes (Clostridium) in winter, of which onlyJanthinobacteriumwas the abundant taxa, the others were the rare taxa. In addition to the genera listed in Fig.2, the rare subcommunity also included 190 genera in spring, 138 genera in summer, 102 genera in autumn, and 141 genera in winter.
The variations in eukaryotic phytoplankton community composition at genus level are shown in Fig.3. The eukaryotic phytoplankton was composed of Chlorophyta (56.87%-98.46%), Bacillariophyta(0.10%-35.99%), Cryptophyta (0.05%-34.27%),and Ochrophyta (0.06%-13.74%). Results demonstrate that Chlorophyta was the major dominant phylum in Fenhe River. The peak of Cryptophyta occurred in winter, and the abundance of Ochrophyta was highest in autumn. At genus level,Desmodesmuswas the dominant genus in the phylum of Chlorophyta, followed byMychonastesof Chlorophyta andCyclotellaof Bacillariophyta in spring, of whichDesmodesmusandMychonasteswere the abundant taxa,Cyclotellawas the rare taxa.In summer,Mychonastes,Golenkinia, andPseudopediastrumwere the abundant taxa, whileDesmodesmus,Messastrum, andMonoraphidiumwith higher relative abundance were the rare taxa.The community was characterized byNannochloropsisandTetraëdronof Chlorophyta, as well as diatomsDiscostellaandStephanodiscusin autumn, of whichStephanodiscuswas the rare taxa.The dominant genera including green algaeMychonastesandMonoraphidiumin winter, all phytoplankton were the rare taxa except forMychonastes. In addition to the genera listed in Fig.3, the rare subcommunity also included 40 genera in spring, 49 genera in summer, 50 genera in autumn, and 33 genera in winter.
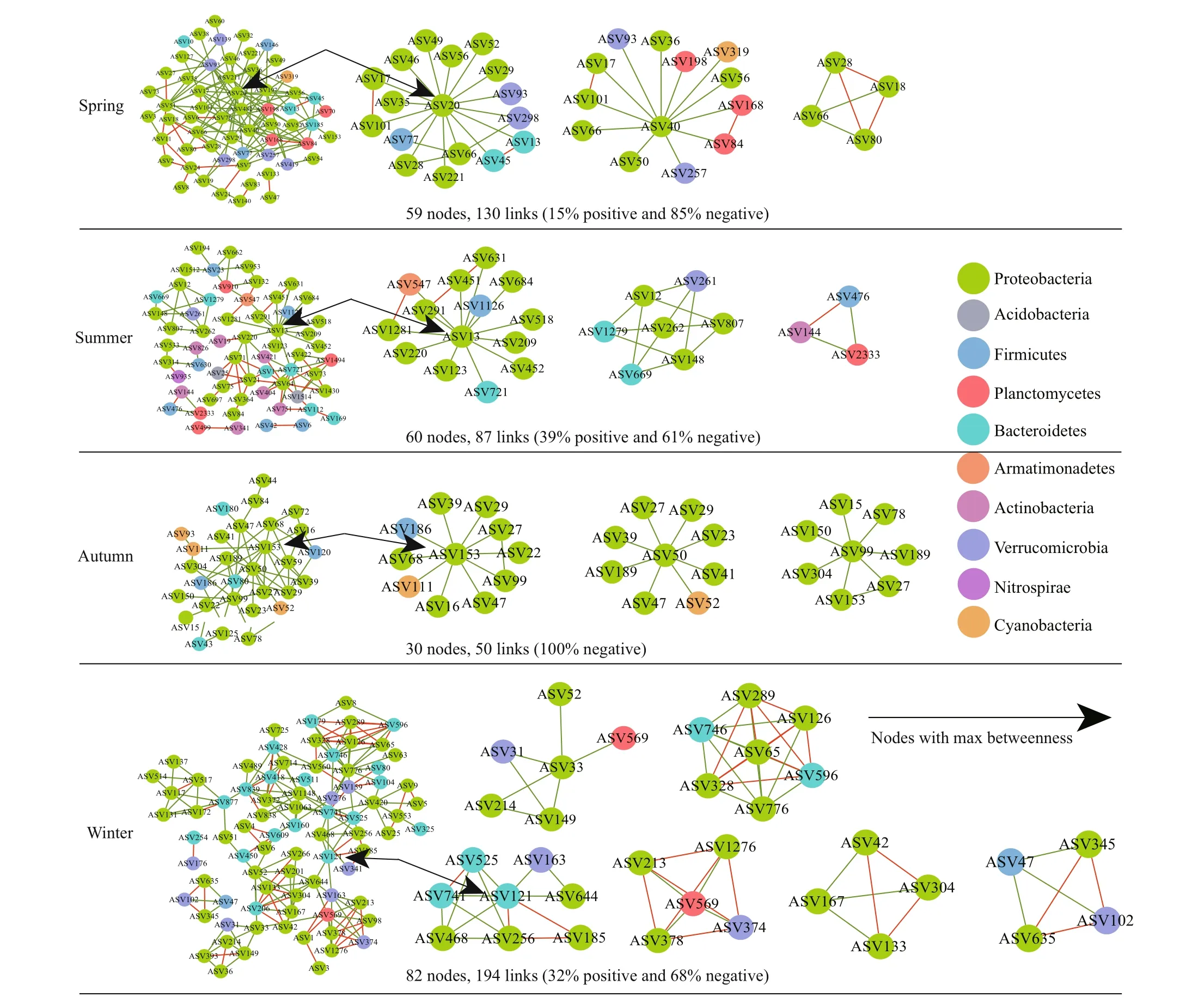
Fig.4 Co-occurrence networks constructed based on bacterial communities
3.4 Co-occurrence networks of bacterial communities
The interaction between diff erent planktons plays a vital role in shaping the distribution pattern of microorganisms. Aquatic ecosystems are composed of networks connecting diff erent organisms of diff erent sizes and are maintained through species-to-species interactions (including symbiosis, competition, and parasitism) (Zhu et al., 2018). Therefore, co-occurrence patterns of each season were dipicted to explore the associations between ASVs, which reflect the potential interactions between microbes in complex communities and ecological processes as well such as historical eff ects, cooperation, and habitat filtering.
The topological properties of bacterial networks over time are presented in Supplementary Table S3.We compared the network topology parameters of the empirical network with the Erdös-Réyni random network, including the modularity index, the average path length, and the average clustering coeffi cient.The results demonstrate that the parameters of the empirical network were significantly higher than the random network, reflecting that the network structure was not randomly distributed. The modularity of all networks was higher than 0.4, demonstrating that networks had a modular structure composed of closely connected nodes and forming a “small world”topology. Overall, the proportion of negative interactions accounted for 68%-100% of the potential correlations observed in four seasons (Fig.4),indicating the co-exclusion (negative interactions,green lines) among taxa is greater than co-occurrence(positive interactions, red lines).
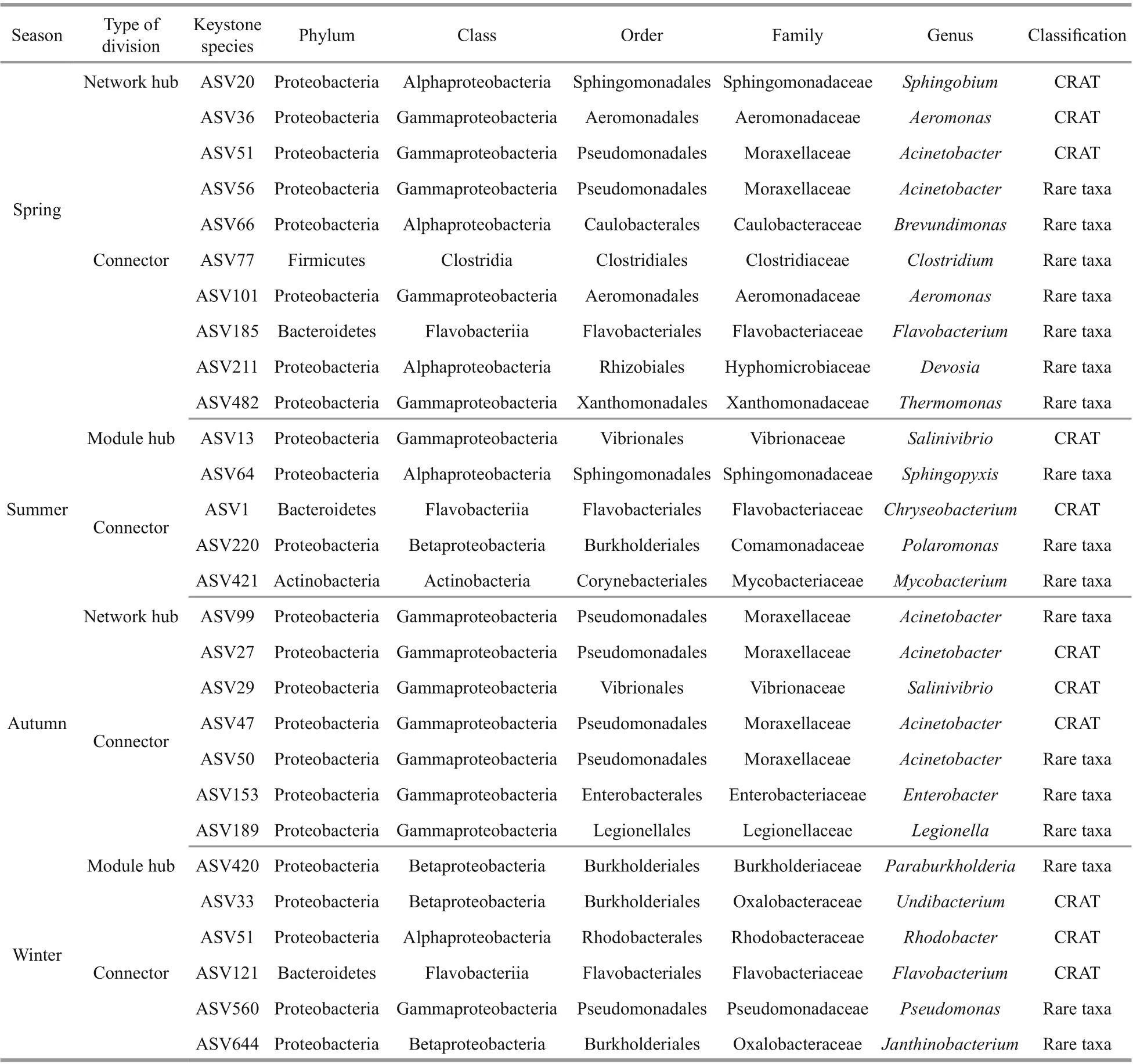
Table 3 Keystone species identified by topological roles of bacterial networks
Among the nodes of bacterial networks,Proteobacteria occupied the largest number of ASVs in all seasons, with 43 ASVs in spring, 32 ASVs in summer, 22 ASVs in autumn, and 53 ASVs in winter.In addition, Proteobacteria occurred in almost all of the sub-network regardless of season (Fig.4). These results demonstrate that some Proteobacteria members can adapt to diff erent ecological environments, and this adaptation could also explain the great abundance of Proteobacteria in all sites in each season. The network is divided into network hubs, module hubs,connectors, and peripherals according to the amongmodule connectivity (Pi) and within-module connectivity (Zi). ASVs that are divided into network hubs, module hubs, and connectors are regarded as keystone species (Supplementary Figs.S1-S2). These nodes can keep the network at the lowest hierarchical structure under the premise of completing its ecological functions (Fan et al., 2018). In order to determine the taxonomic composition of each network, we drew a phylogenetic diagram of the main modules.
Network hubs were detected in spring and autumn only (Table 3). In the spring network, one network hub identified originated from Proteobacteria(Sphingobium) with max betweenness centrality. In addition, nine ASVs were divided into connectors,which were seven Proteobacteria ASVs, one Bacteroidetes ASV (Flavobacterium), and one Firmicutes ASV (Clostridium), respectively.Moreover, three sub-modules were highly correlated.They belong to the rare taxa except ASV36 and ASV51. In addition to Proteobacteria, Verrucomicrobia and Planctomycetes were also predominate in subnetworks. In the summer network, two ASVs were divided into module hubs, belonging to Proteobacteria(SalinivibrioandSphingopyxis), of whichSalinivibrioaffi liated to rare taxa had the highest betweenness centrality; three ASVs were classified as connectors,the genera that could be identified as keystone taxa were mainlyChryseobacterium,Polaromonas, andMycobacterium, of whichPolaromonasandMycobacteriumwere rare taxa. The summer network was relatively diverse, the main components of which were Proteobacteria, Actinobacteria, and Bacteroidetes.The autumn network had fewer nodes and links, the onlyAcinetobactergenus was divided into network hub and six ASVs were classified as connectors.Genera that could be identified as key taxa includedAcinetobacter,Salinivibrio,Enterobacter, andLegionella. In the winter network, one module hub identified was originated from Proteobacteria. And five ASVs were divided into connectors, belonging to Bacteroidetes and Proteobacteria at phylum level. The winter network had the highest modularity. Although the negative correlation accounts for the majority of the entire network, the sub-modules composed of ASV65 (Pseudomonas) and ASV569 (Pirellula)exhibited a high positive correlation (Fig.4).
The interactions between ASVs within phylum or high-level taxa (Supplementary Fig.S3) indicated that taxonomically closely associated bacteria were also ecologically closely related, which in turn also reflected their synergistic relationships or common niche preferences. Apart from the co-occurrence patterns between phyla, such non-random patterns also occurred between diff erent phyla. A typical example was that members of the phylum Proteobacteria showed significant correlations with Bacteroidetes, Planctomycetes, and Actinobacteria.
3.5 Co-occurrence networks of eukaryotic phytoplankton communities
In the network topological properties of eukaryotic phytoplankton (Supplementary Table S3), the average path coeffi cient, average path length, and modularity index in the molecular ecological network were greater than the corresponding values of the random network, indicating that the network constructed in this study conformed to the characteristics of scalefree, small world, and modular, which can be used for the subsequent study of phytoplankton interaction. In spring and summer, the network modularity was relatively higher, and the connections between microorganisms were closer (Fig.5). In the summer and autumn networks, positive correlations accounted for 79% and 59%, negative correlations accounted for 21% and 41%, respectively. On the contrary, in winter,the algal communities almost completely tended to co-exclude, that is, the negative relationships accounted for a higher proportion (92%).
Keystone species identified by topological roles of eukaryotic phytoplankton networks are shown in Table 4. In the spring network, one network hub belonging toScenedesmus(rare taxa) of Chlorophyta was identified. The module hubs that originated fromMychonasteswith the maximal betweenness centrality were identified and the connectors came from Ochrophyta (Nannochloropsis) and Chlorophyta(Choricystis,Pseudoschroederia,Monoraphidium).The spring network presented a co-occurrence pattern between Chlorophyta and Bacillariophyta. ASV16(Mychonastes) was an important hub connecting all modules, forming a sub-network withCyclotella,Pseudopediastrum, andNannochloris. In the summer network,Pseudoschroederiagenus was divided into several module hubs; three ASVs could be divided into connectors, of which two were Chlorophyta and one belonging to Bacillariophyta, the identified genera includingMychonastes,Nannochloris, andCyclotella. As the abundant taxa, ASV39 formed a sub-module withMessastrum,Rotundella,Monoraphidium,Pseudopediastrum, andPseudoschroederia, then indirectly connected with other taxa through ASV95 (Pseudoschroederia). In the autumn network, one module hub was based onCyclotella(Bacillariophyta), including four connectors mainly identified as being originated fromScenedesmus,Monoraphidium,Mychonastes, andFollicularia. Ochrophyta was abundant in autumn and closely related to Chlorophyta in the entire network. In the winter network, six ASVs were divided into several network hubs, of which four were Chlorophyta at the phylum level; and the identified genera includedDesmodesmus,Mychonastes,ASV454Pseudopediastrum, andMonoraphidium,and two ASVs were affi liated toCryptomonasof Cryptophyta. Compared with other seasons, more Cryptophyta (Rhodomonas,Cryptomonas,Rhinomonas,Teleaulax) appeared in the winter network, and these taxa also played an important role in the sub-network. In addition, the co-expression patterns appeared in sub-modules composed of ASV331 (Mychonastes) only.
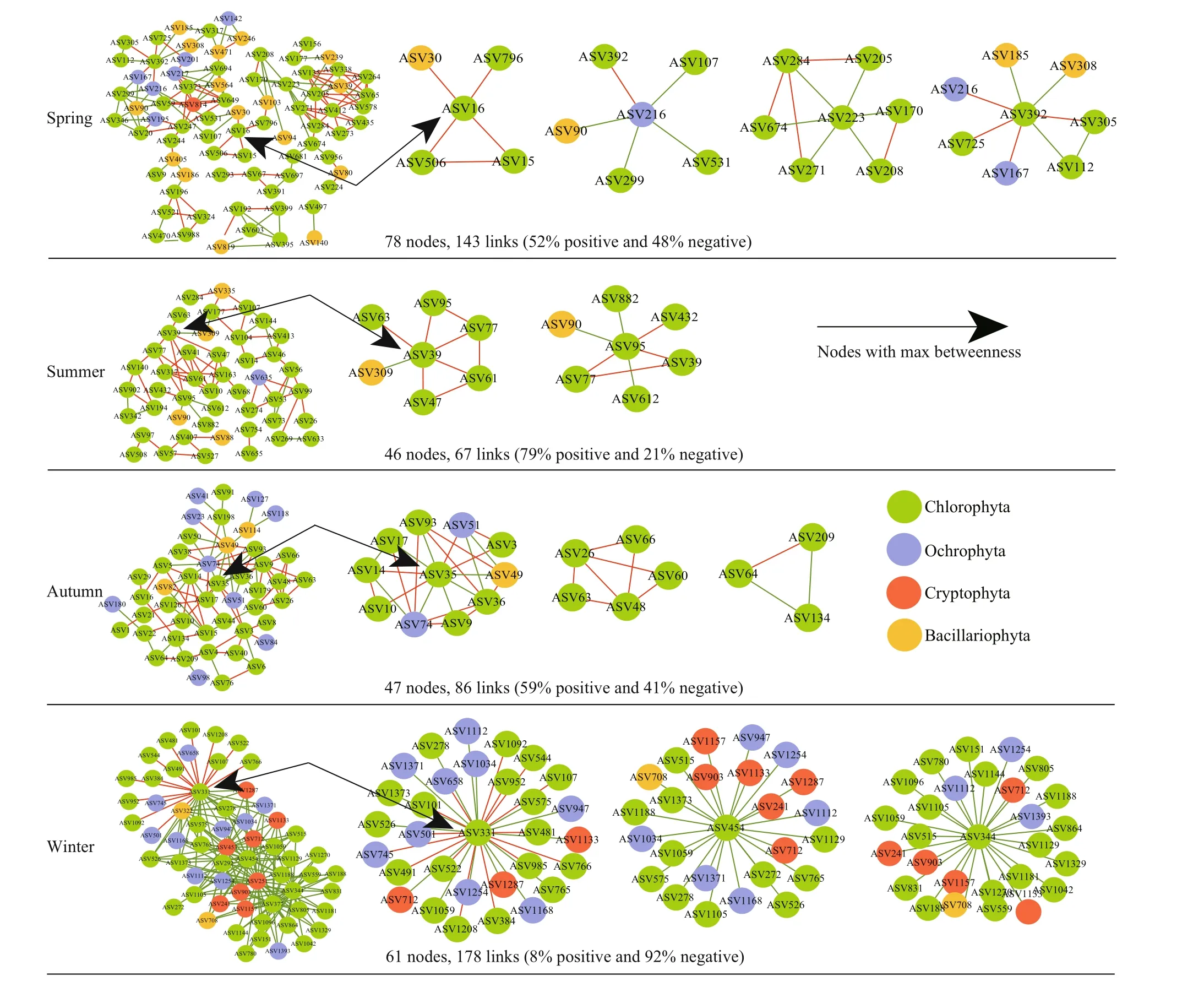
Fig.5 Co-occurrence networks constructed based on eukaryotic phytoplankton communities
The less relevant co-occurrence pattern between ASVs within the phylum was observed (Supplementary Fig.S4), which is consistent with previous observation of taxa over-dispersion (no obvious clustering and correlation across taxa) in plant or animal natural communities (such as sedge community and mammalian community) (Ju et al., 2014). This not only confirmed that over-dispersion of phylogeny may be a common feature of all biological communities from microorganism to macro-plants and animals (Horner-Devine and Bohannan, 2006),but also showed the important influence of negative interaction (such as competition) on community assembly.
3.6 Relationship between keystone species and environmental factors
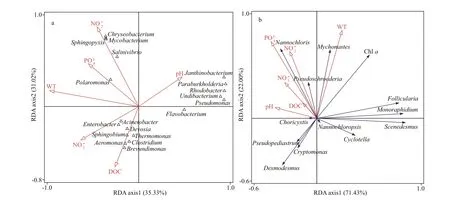
Fig.6 Biplot diagram for CCA/RDA of the relationship between keystone taxa and environmental variables
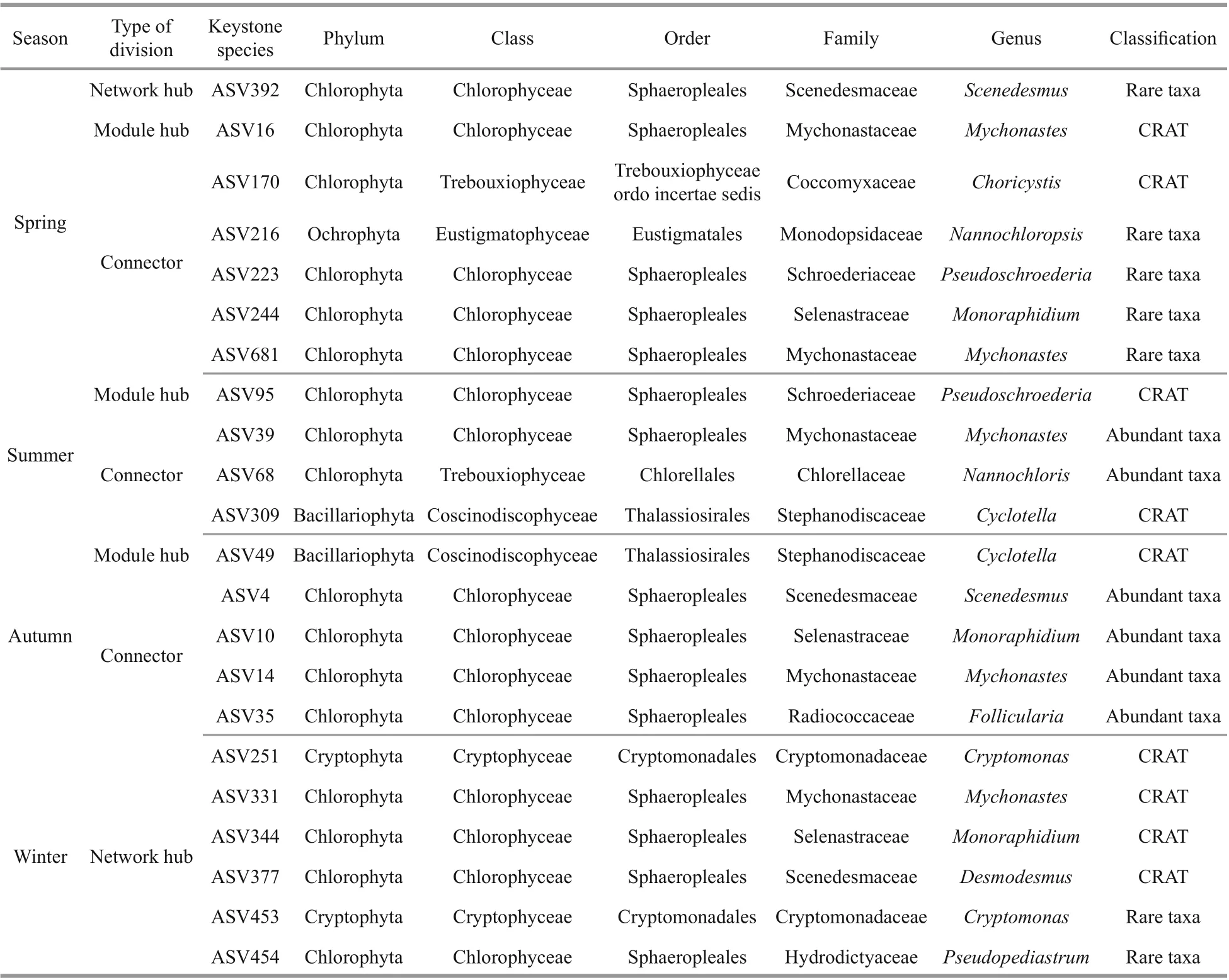
Table 4 Keystone species identified by topological roles of eukaryotic phytoplankton networks
The information of keystone species in bacterial cooccurrence is shown in Table 3. Most of the keystone species detected were affi liated to the rare taxa, and only a small proportion belonging to the conditionally rare and abundant taxa (CRAT). CCA was performed to identify the eff ects of water quality physicochemical variables on these keystone taxa (Fig.6a). The first two axes explained 35.33% and 31.02% of the cumulative variance of the relationship of species-environmental variables, and the eigenvalues were 0.960 3 and 0.843 1, respectively (Supplementary Table S4). The species-environment correlation values of axis 1 and axis 2 were 0.998 8 and 0.979 1, respectively. The Monte Carlo test showed that water temperature,nitrite, and DOC had significant eff ects on the bacterial community (P<0.05).Flavobacterium,Salinivibrio,Mycobacterium,Chryseobacterium, andSphingopyxiswere aff ected by nitrite.Polaromonasshowed significantly positively related to water temperature and phosphate. The most genus of Proteobacteria in spring showed a significant positive correlation with DOC.
The eukaryotic phytoplankton in the freshwater ecosystem responded rapidly and strongly to environmental disturbances and was considered the most important indicator to environmental changes and ecosystem status. Half of the keystone species detected in the eukaryotic phytoplankton cooccurrence patterns were affi liated to the rare taxa,and the rest belonged to the abundant taxa and CRAT(Table 4), indicating that rare taxa were crucial in the aquatic ecosystem. Therefore, RDA was performed to identify the eff ects of water quality physicochemical variables on these keystone taxa (Fig.6b). The first two axes explained 71.43% and 22.00% of the cumulative variance of the relationship of speciesenvironmental variables, and the eigenvalues were 0.454 8 and 0.140 0, respectively (Supplementary Table S4). The species-environment correlation values of axis 1 and axis 2 were 0.874 7 and 0.698 6,respectively. The Monte Carlo test showed that water temperature, nitrate, and phosphate had significant eff ects on the eukaryotic phytoplankton (P<0.05).Water temperature was positively correlated with the abundance ofMychonastes, while negatively correlated with those ofCryptomonas,Desmodesmus,andPseudopediastrum. DOC, nitrogen, and phosphorus (especially nitrate and phosphate) were positively related toNannochlorisandPseudoschroederia,whereas negatively related toCyclotellaandScenedesmus.FolliculariaandMonoraphidiumshowed a negative correlation with pH.
3.7 Co-occurrence patterns between bacterial communities and phytoplankton taxa
Interrelations between bacteria and microalgae are multifaceted and complicated; for example, bacteria can rely on photosynthetic phytoplankton to obtain the organic carbon needed to maintain their growth(Falkowski et al., 2008). In turn, phytoplankton depends on bacteria to remineralize organic matter into inorganic substitutes, ultimately supporting the growth of algae (Worden et al., 2015). The significant interaction between phytoplankton and bacteria has become widely examined in models of, and research on, the microbial loop, particularly in aquatic ecosystems (e.g., Azam et al., 1983; Williams and Ducklow, 2019). Therefore, the research of phycosphere and bacterial communities is important because they control the metabolic interactions of algal-bacteria in the microenvironment. We studied the non-random, co-occurrence networks, and important inter-taxa relationships using the network analysis approach (Fig.7).
In Proteobacteria taxa,Boseain spring,Brevundimonasin autumn, andPseudomonasin winter could promote the growth ofDesmodesmus,whileEnterobacterandOchrobactrumwere mutually exclusive withDesmodesmus.Flavobacteriumwas positively correlated with most green algae, whereas it had a negative correlation withChlorotetraëdronandGoniomonas. The node of green algaeRadiococcushad the highest connectivity in autumn,with 7 positively correlated nodes and 3 negatively nodes. As an abundant bacterium in winter,Janthinobacteriumwas only helpful to the growth of green algaeNannochloropsis.Comamonasbelonging to Comamonadaceae family had a relatively higher abundance in spring and summer, which was considered as denitrifying polyphosphate accumulating microorganism (Calderer et al., 2014),which showed a positive relationship withFolliculariaandPolyedriopsisof green algae.
Spring cyanobacteria had only a negative correlation withSphingobium, and probably promoted the growth of the remaining bacteria (mainly Proteobacteria), Bacillariophyta (Amphora), and green algae (Golenkinia,Phacotus, andNannochloris).Autumn cyanobacteria were more mutually exclusive with Bacteroidetes; specifically,Synechococcuswas negatively related toFlavobacterium. WinterMesorhizobiumwas also the prevalent (negative)group among theSynechococcus-associated bacteria.
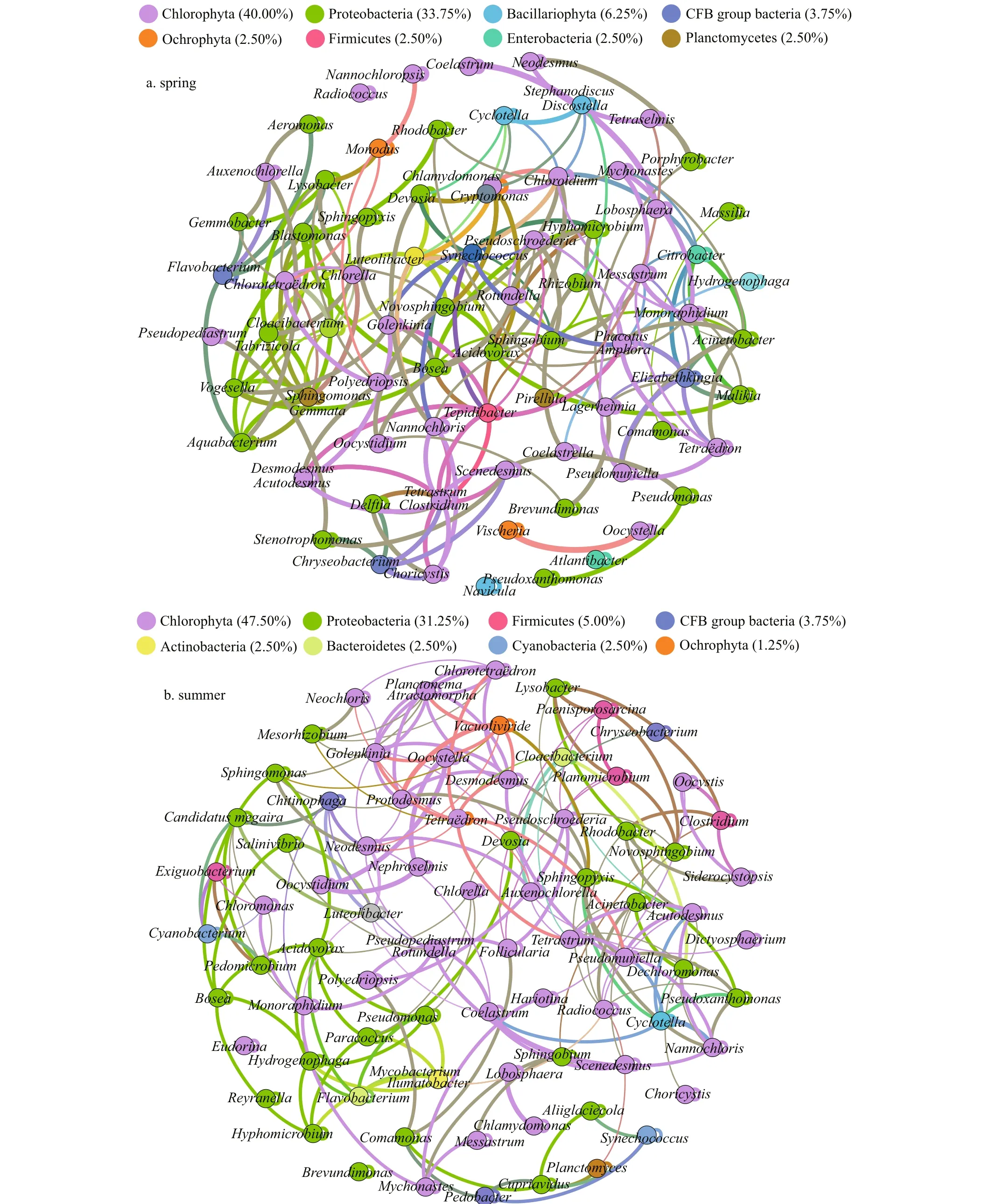
Fig.7 Seasonal co-occurrence patterns between bacteria and eukaryotic phytoplankton at the genus level
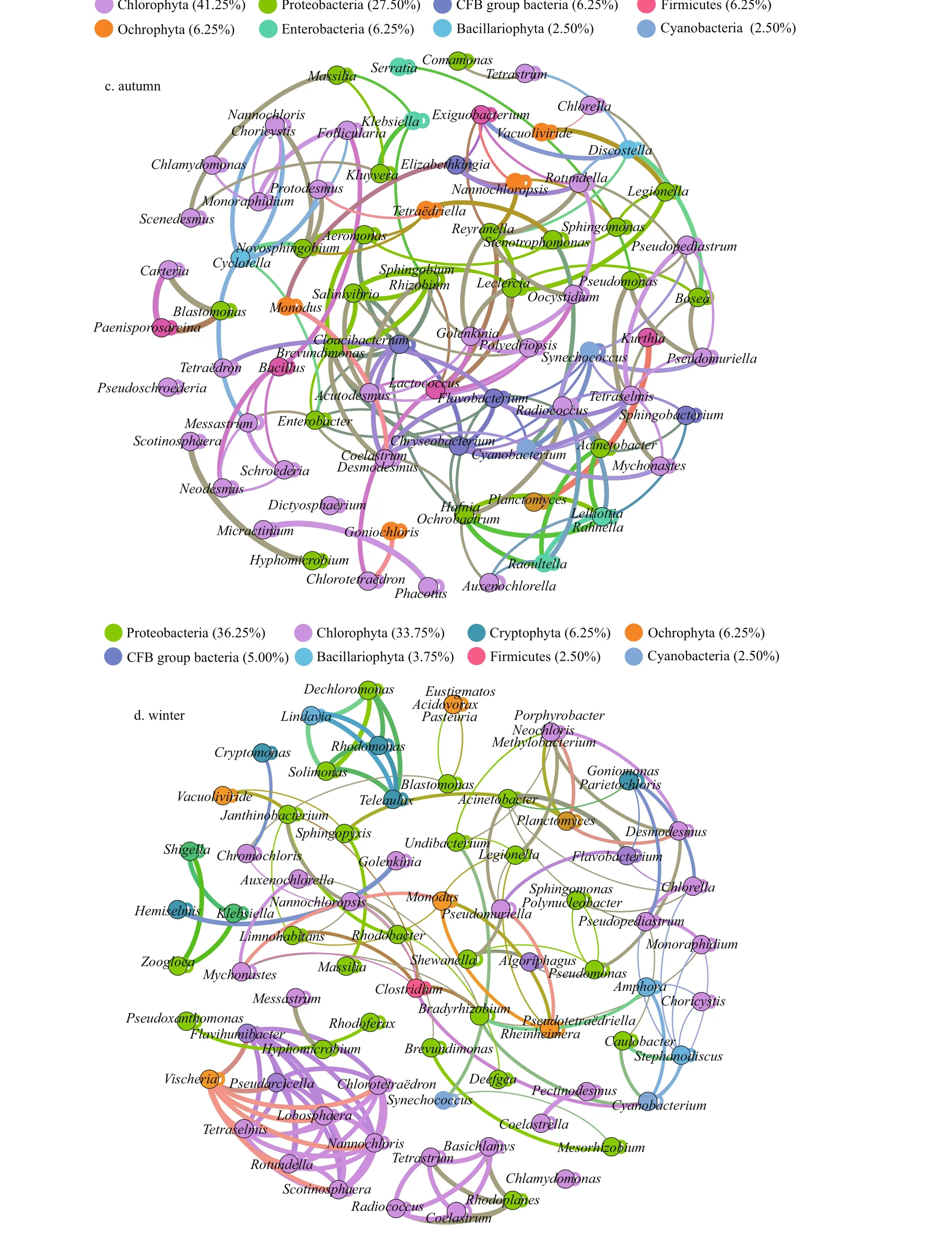
Fig.7 Continued
Discostellais a relatively abundant genus in autumn, the co-existence ofDiscostellaand bacterial Proteobacteria (especiallyReyranellaandBosea)was more conducive to their growth compared to the individual population. The blooms of the small centric diatomStephanodiscuswere regularly found every year in lakes and reservoirs under lowtemperature conditions (Kang et al., 2007). We found thatStephanodiscuswas positively correlated with bacterialCaulobacterandCyanobacterium,whereas negatively related to most green algae in winter. The relationships betweenCyclotellaand bacterial communities did not appear to be onesided,Cyclotellaconstituted symbiotic relationships withSphingopyxis,Novosphingobium, andPseudoxanthomonas. Besides,Cyclotellaalso repelled each other withChlamydomonasandChloroidiumof green algae andCryptomonasof Cryptophyta. The genusAmphorais one of the larger genera belonging to the family Naviculaceae and widespread including freshwater, brackish water,and marine habitats (Nagumo, 2003).Amphorawas positively related to cyanobacteria and most Proteobacteria (Acinetobacter,Hyphomicrobium,andBradyrhizobium), and negatively related toChoricystis.
4 DISCUSSION
4.1 The ecological eff ects of keystone species(rare taxa)
Our results show that Proteobacteria,Actinobacteria, and Bacteroidetes were the dominant phyla of freshwater habitats, which is consistent with previous studies (e.g., Šimek et al., 2005). At the genus level, the seasonality showed a certain trend of change, because some genera dominated in one season while tended to be rare or non-existent in other seasons (Fig.2). The reason might be related to the availability of nutrients and metabolites produced by the rare taxa, which are essential for the reproduction of the abundant taxa (Nyirabuhoro et al., 2020). Another reason might depend on the rarity threshold, below which the dormant groups cannot grow even under favorable conditions (Ruiz-González et al., 2015).
To further determine the contribution and role of abundant and rare taxa, ASVs were classified according to their relative abundance. However, this might cause certain species could not be divided into abundant taxa despite they belonged to a phylum with higher relative abundance. Regardless of this point,we still found that rare taxa were more important in the seasonal dynamics of microbial community because rare taxa contributed more than 93% of bacteria and 63% of eukaryotic phytoplankton, while the contribution rate of abundant taxa was less than 2%. The species rarity of rare taxa may be due to its very narrow environmental niche and is only abundant in a few habitats (Jousset et al., 2017). Besides, rare taxa are generally more sensitive to environmental fluctuations and likely to become extinct because of their low abundance (Zhou et al., 2020). The changing environment is also more likely to influence rare taxa.Therefore, most of the diversity composed of rare taxa may play a critical role in the microbial community composition and dynamics of the Fenhe River. Wang et al. (2020) pointed out that CRAT could switch between abundant and rare taxa under certain environmental conditions and play a key role in microbial community dynamics. Considering that CRAT also accounted for a large proportion in the keystone species of eukaryotic phytoplankton communities, it is of great significance for subsequent research to know the contribution of CRAT to cooccurrence patterns of microbial community.
Moreover, rare taxa are important to the cooccurrence dynamics of microbial community. The interaction between microbial taxa was analyzed through the topological characteristics of cooccurrence network. The co-occurrence network in this study showed the same non-random features and modular structure as the previous studies (Xue et al., 2018; Wang et al., 2020). The number of negative interactions in the seasonal co-occurrence network of bacteria was greater than those of positive interactions, indicating that bacterial taxa tend to compete each other. Among them, most of the negative interactions were observed within rare taxa(spring: 93, summer: 53, autumn: 50, winter: 132),while the interactions between rare and non-rare taxa exist in spring only, indicating that the competition within rare taxa was stronger than that between rare and non-rare taxa. For eukaryotic phytoplankton, the positive interactions within rare taxa (spring: 74, summer: 31, autumn: 6, winter: 15)and between rare and non-rare taxa (only summer:22, autumn: 45) were higher than the negative interactions, indicating that the cooperation within rare taxa and between rare and non-rare taxa was stronger than that within non-rare taxa, which is beneficial to species isolation and modularization of the network (Hu et al., 2017) and maintain the stability of ecosystems. For example, the rareSymbiodiniumtaxa can significantly improve the stability of host-symbiont communities in a changing environment (Ziegler et al., 2018). Besides, the cooperation among these taxa also can contribute to the adaptability of microbial communities in complex environments, because the interaction networks can off er a buff er for resisting environmental disturbance (Konopka et al., 2015).
Rare microorganisms maintain a huge functional gene library, which can enhance indirectly the function of abundant microorganisms (Jousset et al.,2017). As shown in Figs.4-5, most sub-modules were composed mainly of rare taxa, showing the key role of rare taxa in community assembly. Correspondingly,variations in rare taxa also may lead to drastic changes in entire networks and sub-modules (Guimerà and Nunes Amaral, 2005). Degree-based keystone species in co-occurrence networks are often regarded as species that play an irreplaceable role in the microbial communities (Hu et al., 2017). The disappearance or change of these keystone species can lead to the disintegration of modules and networks, even subnetwork (Shi et al., 2016). In this study, there were 24 keystone species belonged to rare taxa, which indicated that rare taxa may have a great impact on other communities (Wang et al., 2020). Thus, we can conclude that rare taxa were more important than abundant taxa in maintaining the composition and structure of entire microbial community.
Nitrogen and phosphorus are key elements for the synthesis of protein and nucleic acid, and they play an important role in the growth, development, and division of cells. Many bacteria populations were significantly correlated with inorganic nitrogen concentrations, such asSalinivibrioandSphingopyxisof Proteobacteria,Chryseobacterium,Mycobacterium,indicating these bacteria groups have potential competition to respond to or use the variations ofinorganic nitrogen in water. This is similar to those Proteobacteria populations that could actively uptake nitrate in coastal surface waters (Morando and Capone, 2016). Besides, the relative abundance of several bacteria populations was significantly related to organic carbon concentrations, includingEnterobacter,Aeromonas, andAcinetobacterbelonging to Proteobacteria. These groups could respond to the organic carbon availability and utilize them as a carbon source. In addition,Polaromonaswas easier to grow in warmer water.
Diff erent nitrogen and phosphorus concentrations also had a greater impact on the growth and physiological and biochemical reactions of algae. It was observed thatNannochlorisandPseudoschroederiahad a positive correlation with nitrate, DOC, and phosphate. However, Shen et al.(2015) showed that compared with organic carbon sources, supply of carbon dioxide under suitable nitrogen and phosphorus nutrient conditions increased the growth rate of microalgae.
4.2 Network associations between bacterial communities and phytoplankton taxa
Although co-occurrence networks can predict the direct associations between taxa, microorganisms may also show positive or negative correlation based on indirect reasons such as environmental preferences(Weiss et al., 2016). The extent to which closely related microbial groups may coexist is still a topic worthy of discussion (Mayfield and Levine, 2010).Previous studies on aquatic microbial communities have demonstrated that closely related taxa have consistent temporal dynamics and similar niches(Andersson et al., 2010).
TheMychonastesare distributed worldwide and have a wide range of habitats, including streams and large still waters, for example, Baringo and Victoria Lakes in Kenya, Erken Lake in Sweden, and Stechlin Lake in Germany, etc. (Krienitz et al., 2011). However,as the most abundant genus in spring, summer, and winter in our study,Mychonasteswas first reported by Li et al. (2013) as a new record genus of freshwater Chlorophyceae isolated from Dianchi Lake.Mychonastesis a taxon that can naturally adapt to the desert climate as its optimal temperature is 30 °C,which is significantly related to its abundance.Synechococcusis a common prokaryotic picocyanobacteria, which had a relatively high abundance in summer. Liu et al. (2019) studied the growth and interspecies competition ofMychonastesandSynechococcusunder diff erent N∶P ratios. Although we have not found the competitive relationship betweenMychonastesandSynechococcus,Mychonasteshad a significant correlation with other cyanobacteria(Cyanobacterium) in autumn.Flavobacteriumis considered as a bacterium that can promote plant growth, which was reported to exist during the algal blooms and can improve the biological phosphorus removal ability, and participate in the cleavage of polysaccharides (Park et al., 2007). In our study, based on positive correlation,Flavobacteriumprobably promoted the growth of most green algae. The node of green algaeRadiococcushad the highest connectivity in autumn. The stability of ecosystem function lies on the interaction between species (Zhou et al., 2010).Lower connectivity in the community will lead to higher functional stability of the system (for example,a non-scale network), because the entire network module is less aff ected by a node loss (Yang et al.,2017).
Cyanobacteria are the only prokaryotes with oxygen-producing photosynthesis. They exist in diff erent niches and are important participants in the global carbon and nitrogen cycle. The planktonic cyanobacteriumSynechococcusis ubiquitous in the ocean and fresh waters and plays an important role in total carbon sequestration on a global scale in oligotrophic aquatic environments (Callieri et al.,2012). We observed thatSynechococcuswere negatively related to other bacteria, the negative correlations in co-occurrence patterns might imply the predation or competition among taxa. The presence of correlations between populations increases the probability that these populations interact. However, the relationship of these populations may change when diff erent populations are influenced by the same factor. Therefore, not all algae-bacteria connections can be identified by correlation methods, and correlations may not necessarily come from the connections between populations (Paver et al., 2013), which may also be indirectly aff ected by the external environment.
Numerous pieces of evidence have been demonstrated that a link occurs between diatoms and microorganisms (Behringer et al., 2018); however,whether they cooperate or exclude each other still lacks relevant knowledge. In our results, we found significant correlations between diatom communities and bacteria communities. In addition, these relationships were greatly subjected to seasonal variations. There is considerable evidence that the combination of diatoms and bacteria is beneficial to the growth of algae (Grossart et al., 2005) and bacteria(Stanish et al., 2013). The symbiotic relationships between Bacillariophyta and phylogenetically related bacteria verified that cell yield and growth rate of bacteria increased when grown in mixed culture,because the organic material secreted by diatoms was absorbed by and would support the growth of bacteria(Jolley and Jones, 1977).
The growth stages of plankton throughout the year include mainly: winter (transition stage, December-January), spring (accumulation stage: March-April;climax transition stage: May), summer (water bloom outbreak stage: June-August, depletion stage:September-November). Diff erent microorganisms may play the same ecological role over time, which may explain the unique keystone taxa detected in diff erent ecological networks. Therefore, our results provide evidence for the profound influence of physico-chemical conditions on the seasonal dynamics of microbial community composition.
5 CONCLUSION
The eukaryotic phytoplankton and bacterial networks constructed in our research conformed to the properties of having non-randomly connections,modular structure, and “small world”. In cooccurrence patterns, the positive interactions between bacteria and eukaryotic phytoplankton ASVs may be evidence of mutual influence (cooperation), while the negative correlations may indicate predation or competition. Through its within-module connectivity(Zi) and among-module connectivity (Pi), we identified the keystone species that can maintain the stability of microbial networks. Most of the keystone species were defined as rare taxa, a few abundant taxa were still observed although rare taxa accounted for a large proportion. In the bacterial networks, ASVs of Proteobacteria (Aeromonas,Devosia,Salinivibrio,Sphingopyxis, andAcinetobacter) and Acidobacteria(Mycobacterium) can play an important role in the stability of ecosystem; and in eukaryotic phytoplankton networks, ASVs of Chlorophyta (Mychonastes,Pseudoschroederia,Follicularia, andMonoraphidium), Bacillariophyta (Cyclotella), and Cryptophyta (Cryptomonas) were significantly related to water temperature and inorganic nitrogen.With the disappearance of the “keystone species” in the network center, the aggregation structure will suff er major damage, which may bring huge biogeochemical consequences. The abundance of these keystone species was significantly related to many ecosystem functions and environmental changes, highlighting the key ecological role of rare species, and providing a new ecological significance for the seasonal co-occurrence patterns of bacteria and eukaryotic phytoplankton in urban aquatic ecosystems.
6 DATA AVAILABILITY STATEMENT
The datasets generated and/or analyzed during the current study are available in the Sequence Read Archive (SRA) of NCBI repository, under accession number SRP273360.
 Journal of Oceanology and Limnology2022年4期
Journal of Oceanology and Limnology2022年4期
- Journal of Oceanology and Limnology的其它文章
- Validation and error analysis of wave-modified ocean surface currents in the northwestern Pacific Ocean*
- The energy conversion rates from eddies and mean flow into internal lee waves in the global ocean*
- Observation of physical oceanography at the Y3 seamount(Yap Arc) in winter 2014*
- Decadal variation and trend of the upper layer salinity in the South China Sea from 1960 to 2010*
- Observations of turbulent mixing and vertical diff usive salt flux in the Changjiang Diluted Water*
- Experimental research on oil film thickness and its microwave scattering during emulsification*