
先兆子痫及胎儿宫内发育迟缓的胎盘共表达模块分析及基因富集分析
2022-08-12 05:53刘福林王宝峰
武汉大学学报(医学版) 2022年2期
夏 慧 叶 尚 刘福林 王宝峰,4
1深圳出入境边防检查总站医院 广东 深圳 518013;2深圳市南山区蛇口人民医院 广东 深圳 518067;3法国国家健康与医学研究院附属于巴黎大学 法国 巴黎 75006;4鄂尔多斯妇产医院妇产科 内蒙古 鄂尔多斯 017000
先兆子痫(pre-eclampsia,PE)往往伴随于妊娠期高血压而发生,是妊娠期高血压疾病的常见并发症,其全球范围内的发病率约为2%~8%,而其所致的孕产妇死亡率占比高达14%[1,2]。研究表明胎盘滋养细胞在母体子宫内膜的异常植入可导致子宫螺旋动脉的重建异常而导致母体与胎儿血液在进入胎盘内流动时发生不畅而引起母体血压增高,从而导致PE的发生[3]。同样地,胎盘发育异常也可胎儿宫内发育迟缓(intrauterine growth restriction,IUGR),其与PE同属于严重妊娠疾病,表现为胎盘因素等所致的胎儿在宫内的生长发育受限,导致胎儿的健康及生命遭受严重威胁[4,5]。然 而,无论是PE还是IUGR,尽管已有大量研究,但其具体发生机制尚不十分清楚。
本研究利用加权基因共表达网络分析(weighted correlation network analysis,WGCNA)分析了由Awamleh等[6]上传的关于正常胎盘、PE胎盘、IUGR胎盘及PE+IUGR胎盘组织的基因测序数据集,从更宏观的角度探讨了在PE及IUGR发生中起重要作用的基因集,并且本研究还系统比较了主要富集通路在两种疾病中所起的作用及其差别。本研究所运用的WGCNA方法已证实广泛运用于分析高通量数据集,在寻找基因间的关联性及构建共表达网络上具有重要作用[7]。通过新方法的运用,本研究旨在为PE及IUGR的研究提供更广阔的思路。
1 资料与方法
1.1 高通量数据集的收集进入NCBI的GEO数据库(http://www.ncbi.nlm.nih.gov/gds/),并下载GSE114691。GSE114691由Awamleh等[6]上传,其来源于患有或不患有PE及IUGR的患者,包括21个无疾病对照组胎盘组织,20个PE胎盘组织,18个IUGR胎盘组织以及20个PE及IUGR混合并发症的胎盘组织。从组织内分离RNA后由Illumina HiSeq 2000平台进行检测。
1.2 WGCNA分析运用R软件(version 3.60)中的Affy包(version 1.64)对原始数据进行预处理,包括读取原始数据,归一化矫正,去除包含缺失值的行。构建以患有或不患有PE及IUGR的表型文件。将所有不包含缺失值的行纳入WGCNA分析,本研究中共纳入12 084个基因。运用WGCNA包(version 1.67)对样本进行聚类以剔除离群样本,筛选软阈值进行网络构建,将邻接矩阵转换为拓扑重叠矩阵用于筛选相关模块并以不同颜色表示,构建基因与性状散点图并绘制相关性热图。
1.3 关键基因的富集分析及比较提取相关系数(r)大于0.4以上的彩色模块中的基因进行基因本体论(gene ontology,GO)富集分析和京都基因与基因组百科全书通路(Kyoto encyclopedia of genes and genomes,KEGG)分析。分析的可视化由R包clusterProfiler(version 3.14)执行并且以P<0.05作为筛选标准。最后,比较对照组、PE组、IUGR组、PE&IUGR组基因富集分析的结果。
2 结果
2.1 样本聚类及性状匹配下载原始数据,并用Affy包进行原始数据整理。通过排除缺失值以满足WGCNA算法分析模式。最后纳入12 084个基因用于后续分析。对不同组别的样本表达矩阵进行组合,构建WGCNA分析所需要的矩阵形式,同时构建样本表型文件。对对照组、PE组、IUGR组及PE&IUGR组的样本进行K-means方式聚类,同时对样本所属性状进行了匹配。结果显示IUGR5样本明显偏离群体,故予以剔除后再进行分析。
2.2 软阈值筛选及共表达模块构建以及模块与基因相关性热图运用WGCNA计算共表达网络相关性首先需要确定用于分析的软阈值,本研究设定1~30范围进行分析,当r2大于0.9时则选取用于后续分析。本例中选取的软阈值为22。同样的,本研究成功获取多种颜色共表达模块,并且对模块进行了合并处理以剔除只包含少量基因的共表达模块。最后,本研究通过热图呈现了基因与模块间的相关系数,其中较相关区域(浅黄色)提示了关键模块与基因的存在(图1)。

图1 共表达模块构建及合并后模块中基因的相关系数热图
2.3 共表达模块筛选与基因性状系数散点图运用WGCNA分析性状表型与不同模块之间的关系,可得到如下所示的相关系数热图(图2A)。结果显示多数模块与正常对照组相关,少数模块与其它疾病组具有相对较低的相关性。接着提取不同模块中的基因与性状的相关系数作散点图,下图只展示部分代表性模块。结果显示,在PE组蓝色模块中,有323个基因与此性状具有相关性,相关系数较高(r=0.42,P<0.05);在IUGR组灰色模块中,318个基因与性状具有相关性,相关系数较高(r=0.4,P<0.05)。本研究保留PE组(图2B)及IUGR组(图2C)有意义模块中的基因进行后续分析及比较。

图2 共表达模块与性状相关系数热图及基因性状相关系数散点图
2.4 PE及IUGR组关键模块中基因富集分析及比较纳入PE组及IUGR组关键模块中基因用于富集分析。本研究采纳了GO分析中的生物过程(biological process,BP)分析,以及KEGG的信号通路(Pathway)分析,展示了富集程度最显著的15个词条。结果显示,PE组中GO_BP分析提示了蛋白合成及折叠、DNA转录、DNA损伤调节、细胞周期等的异常(图3A),同时KEGG信号通路提示了RNA降解、剪接体、脂质代谢及激素合成等方面的异常(图3B)。IUGR组中GO_BP分析提示了甾体类激素合成、细胞黏附、免疫反应等方面的异常(图3C),同时KEGG信号通路提示了补体反应、氨基酸代谢、糖代谢、磷酸化反应、p53信号通路等方面的异常(图3D)。同时本研究比较了PE组及IUGR组富集词条,结果显示,GO_BP分析在两组中具有较大差异,无具有统计学意义的词条重复(图3E);而KEGG通路提示除甲状旁腺激素代谢及寿命调节通路的重叠外,无其它具有统计学意义的词条重复(图3F)。这些结果反映了本研究筛选基因及其对应通路的特异性。
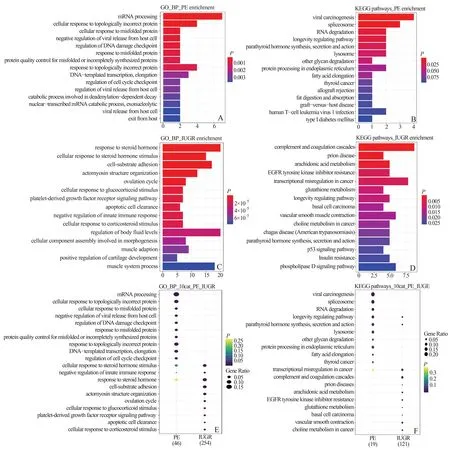
图3 PE组及IUGR组关键基因的富集分析及比较
3 讨论
PE及IUGR在致孕产妇及胎儿死亡方面具有高危性,而其相关基因及其通路的研究仍不明了,既往关于PE及IUGR的研究,以本例所下载数据集的相关文献为例,一般采用优化t检验检测不同组别之间的基因表达并筛选出最具表达差异的基因集,随后对这些数据集进行富集分析。然而,单纯的差异基因表达检测忽略了基因表达在生物体内的相关性,割裂了基因表达的整体性,片面地富集最高差异表达的基因会丧失更全面的通路信息,而WGCNA通过计算相关性则尤其适用于分析具有n×m矩阵形式的临床样本微列阵数据[7]。
WGCNA包被广泛用于构建共表达网状,分析基因与性状之间的关系,探索有意义的表达模块,计算相关性,可视化等,其近年来尤其用于探索肿瘤基因表达与临床表征之间的关系,然而WGCNA鲜少用于探索研究人母胎临床表征及疾病之间的关系。既往文献仅涉及运用WGCNA探索全基因组表达与母体体重指数及胎儿生长之间的关系,以及用于发现与妊娠期糖尿病相关的潜在生物学标志物[8-10]。目前尚无研究运用WGCNA探索PE及IUGR的特异基因及相关通路。因此,本研究旨在通过WGCNA法分析PE及IUGR的特异基因及相关通路。值得注意的是,虽然本研究并无一般研究中的临床特征指标(如年龄、体质量等),但本研究创意性地将正常、PE、IUGR、PE&IUGR作为了样本的临床表型进行相关性分析。
本研究剔除了IUGR第5个样本,因聚类分析结果显示其为离群值,从样本聚类结果来看,样本聚类结果较为紧密,适用于后续分析。后续分析结果提示大量基因与正常对照组相关,而少量与PE或IUGR有关。鉴于导致疾病发生的基因或参与通路可能占总体基因的比例较小,大部分的基因整体表达趋于正常,此结果则可能是合理的。而PE&IUG联合组未能检测到大量较高相关性的基因则可能归因于其各自特异性的基因及通路较少重合,从而形成数据噪音,影响了对潜在有效基因的检测,降低了基因与该性状的相关程度。本研究的富集分析结果提示,PE与细胞生命周期有关,这与既往关于胎盘细胞衰老在胎盘中作用的研究[11]一致;同时PE发生伴随的DNA转录过程及蛋白质合成及代谢异常也见诸于既往的报道[12]之中。IUGR的发生及参与的甾体类激素合成、氨基酸代谢、p53信号通路等同样在既往研究[13-15]中有报道。故而说明本研究中可能存在尚未经其他研究提及的有用通路,而这些通路可能具有进一步的研究价值。此外,本研究还通过基因与通路的热图展示了参与相关通路的相应基因群。例如在IUGR组中,热 图 显 示CFLAR、CRY 2、FBXO32、FOXO1、FOXO3、KLF9参与了细胞受糖皮质激素刺激的反应过程,这既与之前的系列研究[13,16]所得的糖皮质激素参与IUGR发生的结论一致,又弥补了既往研究在探究其中具体基因方面的缺陷。
因此,本研究通过新方法WGCNA的运用,分析了PE及IUGR组临床样本的微列阵数据,揭示了PE及IUGR的发生发展过程中所涉及到的各自特异性的参与基因及相应信号通路,为进一步研究PE及IUGR的胎盘发生机制提供了新思路。
猜你喜欢
作物学报(2022年1期)2022-11-05
中国现代医生(2022年19期)2022-11-04
中国医学物理学杂志(2022年9期)2022-10-09
水产养殖(2022年8期)2022-09-21
广东农业科学(2022年7期)2022-09-14
中国现代医生(2022年19期)2022-08-25
现代仪器与医疗(2021年6期)2022-01-18
家庭百事通·健康一点通(2016年9期)2016-09-21
大众健康(2015年2期)2015-09-10
作文与考试·高中版(2008年11期)2008-11-21
