
空肠弯曲杆菌诱导的结直肠癌的转录组学分析
2022-08-11 14:36李燕毛玉丹张幸鼎牟相宇
中山大学学报(医学科学版) 2022年4期
李燕,毛玉丹,张幸鼎,牟相宇
(中山大学医学院,广东深圳 518107)
结直肠癌(colorectal cancer,CRC)发病率在癌症中居第三位,其发生与肠道微生物有着密切关系。已有研究发现,拟杆菌属(Bacteroides)、梭杆菌属(Fusobacterium)、沙门氏菌属(Salmonella)、埃希氏杆菌属(Escherichia)和弯曲杆菌属(Campylobacter)等多种肠道病原体与结肠癌的发生发展密切相关[1-4]。空肠弯曲杆菌(Campylobacter jejuni,C.jejuni)是一种带荚膜的革兰氏阴性杆菌,是常见的人畜共患食源性病原菌之一。人类感染C.jejuni后,会产生肠胃炎、腹泻、腹部绞痛以及发热的症状,并可能会伴有反应性关节炎、肝炎、格林-巴利综合征和瑞特氏病等免疫性损伤性疾病[5-6]。C.jejuni81-176 会通过产生细胞致死性膨胀毒素进而诱导DNA 损伤,在小鼠模型中促进结直肠癌的发生[7]。因此,C.jejuni感染不仅会导致患者产生肠胃炎,还有增加患者罹患结直肠癌风险的可能。然而,在C.jejuni诱导的结直肠癌中,宿主基因是如何参与癌症发生发展的,这一点仍不清楚。转录组学测序(RNA sequencing,RNA-seq)技术可以从整体水平上反映细胞中基因转录情况[8]。为了更好的了解空肠弯曲菌诱导的结直肠癌中的基因表达情况,本研究使用了C.jejuni诱导的ApcMin/+小鼠结直肠癌模型,在试验终点分离小鼠结直肠肿瘤组织以及癌旁组织,分别提取组织RNA 后进行转录组学测序分析,探索、筛选出差异基因,以提高对C.jejuni致癌的作用机制的认识,为结直肠癌的防治提供新的探索方向。
1 材料与方法
1.1 C.jejuni 81-176的培养
人类临床分离株C.jejuni81-176 用弯曲杆菌属选择性培养基在37 ℃下微需氧条件下培养48 h。
1.2 动物实验
本实验共选用18 只体质量约为(18.0±3.0)g,雌性C57 BL∕6ApcMin/+小鼠(5-8 周龄),小鼠购买自江苏集萃药康公司,于中国科学院广州生物医药与健康研究院动物房SPF 级环境中分笼饲养,实验经过中国科学院广州生物医药与健康研究院实验动物福利与伦理委员会批准(伦理编号:N2022003)。随机分为两组,计为Mock(空白对照组,n=9)和Campy(C.jejuni处理组,n=9)。分别放入4 笼小鼠笼中(每笼4~5只)。
检疫合格的小鼠在动物房观察一周后,进行打耳标标记。为了使菌在肠道内更容易定植,第1~7天,给小鼠饮用含有4 种混合抗生素的饮用水(100 mg∕L 万古霉素、200 mg∕L 甲硝唑、200 mg∕L 氨苄青霉素和200 mg∕L 新霉素)。抗生素处理后,第8 天以1 × 108CFU∕只的量给Campy 组小鼠灌胃C.jejuni,Mock 组以等量PBS 作为对照灌胃。随后,在第15~20 天,在饮用水中加入25 g∕L 的葡聚糖硫酸钠(DSS)。在第60 天,安乐死小鼠,并进行实验材料取材。关于肿瘤部位与癌旁组织的取材实验时是以肉眼可见的隆起为肿瘤组织部位,以肿瘤中心为圆心距离3 mm 的地方为癌旁,癌旁组织所取的面积约为0.3 cm× 0.3 cm。肠道组织HE 染色由赛维尔生物科技有限公司完成。因Mock 组一只小鼠在予以DSS 水处理期间发生死亡,本文选用Mock组3只小鼠和Campy组4只小鼠的肿瘤和癌旁部位进行转录组测序。
1.3 RNA提取及文库构建与高通量测序
分别取对照组(3 个生物学重复)和实验组小鼠(4 个生物学重复)的结肠肿瘤和癌旁组织,放入装有1 mL RNA later 的2 mL 离心管中,液氮速冻后转移至-80 ℃超低温冰箱保存。提取RNA 时,先向2 mL EP 管中加入1.5 mL TRIzol 裂解液,后取适量组织于液氮保护下将组织样品快速研磨成粉末,转入裂解液,使组织细胞充分裂解。按Invitrogen TRIzol 试剂盒(赛默飞有限公司)说明分别提取组织的总RNA,Fragment analyzer(安捷伦生物有限公司)对RNA 的质量和浓度进行评估,质量检测合格(28 S∕18 S >1.0,RQN 值>7.0)的样本用Promega反转录试剂盒(普洛麦格生物技术有限公司)进行反转录(操作步骤按照说明书进行),按照SMART cDNA 文库构建试剂盒(宝生物工程有限公司)说明书分别对样本进行文库构建。构建好的文库在BGISEQ 测序平台(华大基因,中国深圳)进行转录组测序。
1.4 测序数据过滤及参考基因组比对
本实验委托华大基因进行,具体步骤如下:利用过滤软件SOA Pnuke 软件[9]过滤低质量reads,即去除测序片段中包含接头的reads、去除未知碱基N含量大于5%的reads、去除低质量(质量值低于15的碱基占该reads 总碱基数的比例大于20%)的reads,筛选获得clean reads,利用HISAT2 2.0.4 软件[10]将clean reads 比对到参考基因组序列,使用Bowtie2 软件[11]将clean reads 比对到参考基因序列上得到比对结果。比对完,通过统计比对率、reads在参考序列上的分布情况等,判断比对结果是否通过第二次质控(QC of alignment)。若通过,则进行后续的数据分析。使用RSEM 软件[12]计算各个样品的基因表达水平。
1.5 生物信息学分析
进行基因定量分析、基于基因表达水平的各项分析(主成分、相关性、差异基因筛选等等),并对筛选出的样品间差异表达基因通过DAVID 平台进行gene ontology(GO)富集分析和KEGG通路显著性富集分析等更深入的挖掘分析。
参考物种信息如下,物种名:Mus_musculu,参考基因组版本GCF_000001635.26_GRCm38.p6(NCBI)。
1.6 qRT-PCR验证
根据反转录试剂盒(宝生物工程有限公司)进行反转录获取cDNA,用qPCR 验证筛选出的差异表达基因。反应条件为95 ℃30 s,95 ℃10 s,60 ℃30 s,40 个循 环。采用2-ΔΔCt法分析mRNA 表达 水平。引物由擎科生物科技有限公司合成,详见二维码1。
1.7 统计学处理
使用GraphPad Prism 8.0 和SPSS 25.0 软件处理。小鼠肠道肿瘤的数目的统计学差异比较经KS 检验数据正态分布和单因素ANOVA 分析中的方差齐性检验方差齐性后,使用双尾未配对t检验计算。差异基因的筛选和后续分析基于R 软件中的DESeq2 程序包进行分析,利用Benjamini 法校正后的P值以控制错误发现率,校正后的P值<0.05 以及|log2fold change|>2作为显著差异表达的阈值。
2 结果
2.1 C.jejuni诱导小鼠结直肠癌造模情况
如图1 所示,Campy 组小鼠的成瘤数量比Mock组小鼠的成瘤数显著性增多,差异具有统计学意义,统计数据两组均符合数据正态分布和方差齐性,经独立样本t检验,(t=-3.079,P=0.008 <0.05)。D 图展示的肠道HE 染色结果,Campy 组的肠道黏膜层增厚,中央见坏死区,上皮细胞边缘不清,细胞核浆比增大,见不少核染色深,核仁明显,有见核分裂象。Mock 组相较之下结肠腺体排列规则,细胞核比例大小正常。

图1 C.jejuni促进小鼠肿瘤发生Fig.1 C.jejuni promotes tumorigenesis in mice
2.2 转录组测序数据质量情况
2.2.1 测序数据质控概况 每个实验小组小鼠的结直肠肿瘤以及相对应的癌旁组织样本进行转录组分析,本项目使用BGISEQ 平台一共测了14个样品,每个样品平均产出6.67G 数据。样品比对基因组的平均比对率为90.89%,比对基因的平均比对率为68.71%,一共检测到18 964 个基因。测序质量值大于30(Q30)的碱基占所有碱基的89.52%以上,测序比对效率均值约在90.25%。测序数据质量良好,满足后续分析要求(表1)。

表1 小鼠结直肠样本RNA-seq测序数据质量Table 1 Quality of RNA-seq data from mouse colorectal samples
2.2.2 基因表达水平分布 经过预处理和过滤后,基因表达量用FPKM(Fragments Per Kilobase of exon model per Million mapped fragments)值归一化表示,即每千个碱基的转录每百万映射读取的碎片大小,是对基因长度和测序深度进行校正后的值。本研究使用箱线图展示各样品基因表达水平的分布情况,评估各样本中基因表达数据是否具有对称性,分布的分散程度等信息。如图2 所示,基因表达于各样本中差异不大,分散程度相似。
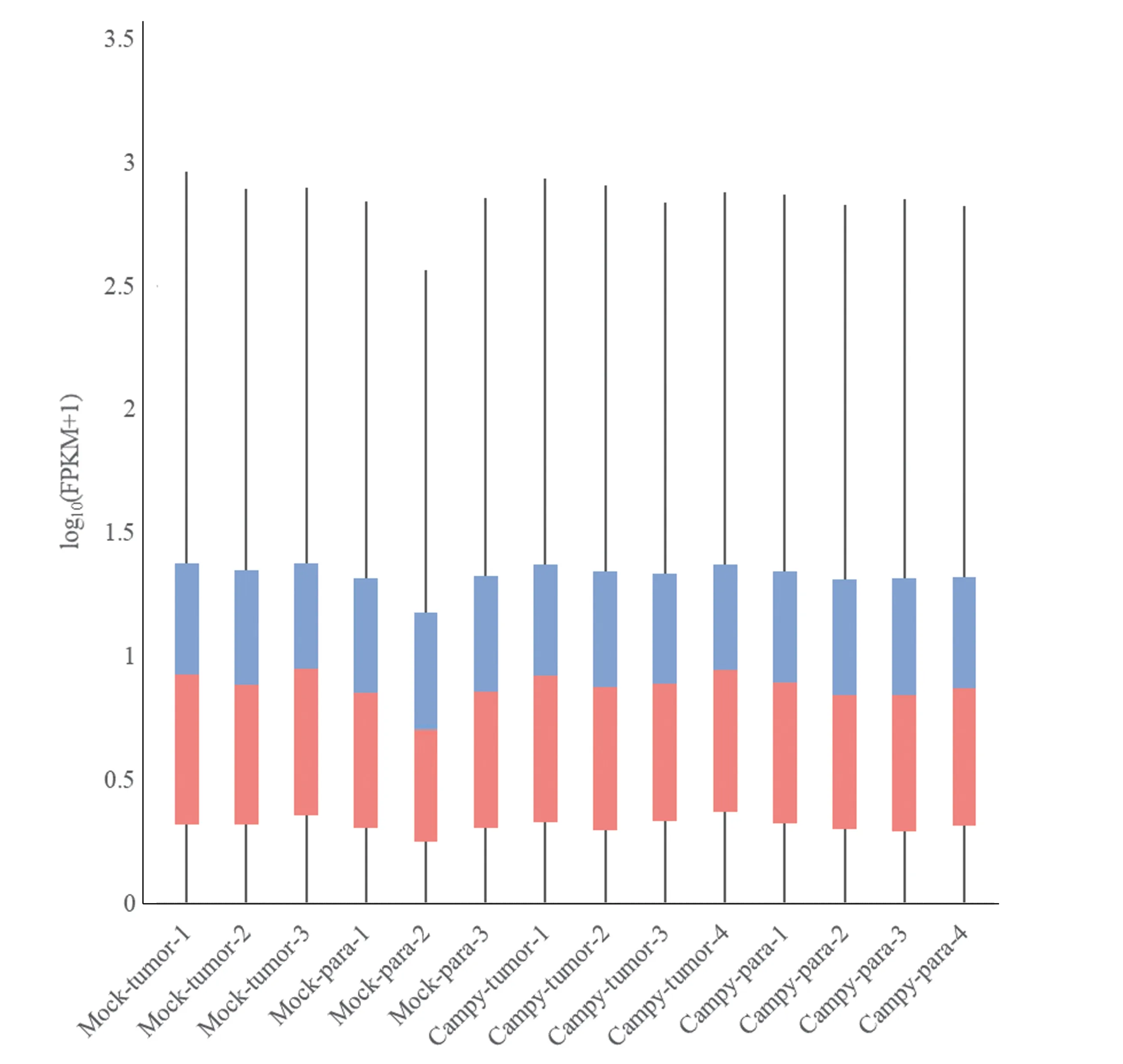
图2 样品基因表达水平分布图Fig.2 Distribution of gene expression in various samples
2.3 基因表达差异分析
经DEseq2 分析,本试验在筛选的过程中,将差异倍数(fold change,FC)的绝对值>4,即|log2FC|>2,P值<0.05 作为筛选标准。通过热图可以发现,经过聚类,同一组内表达相似的基因聚类后几乎都出现在同一簇中。其中,FC 表示的是两个组之间基因表达量的比值。差异表达基因(differentially expressed genes,DEGs)分布情况用火山图表示。如图3 所示,通过测序结果显示,比较Campy 组肿瘤和癌旁组织,即Campy-tumor 与Campy-para,共发现394个显著差异基因,其中341个基因上调,53个基因下调。比较Campy-tumor 与Mock-tumor,共有501个显著差异基因,其中126个基因表达上调,375 个基因表达下调。比较Campy-para 与Mockpara,共有316 个显著差异基因,其中上调基因215个,下调基因101个。
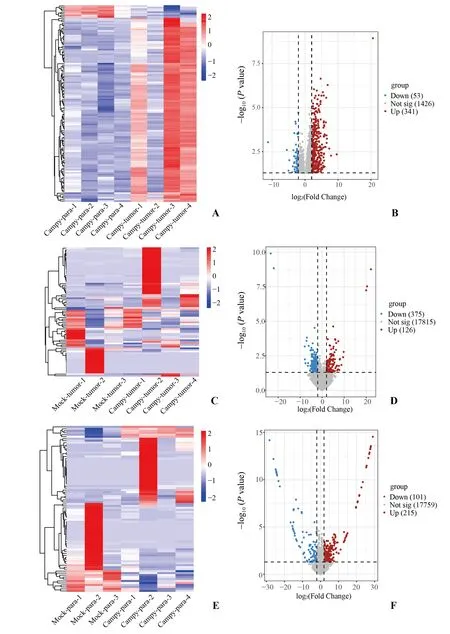
图3 样本间基因表达差异分析Fig.3 Analysis of gene expression differences between samples
2.4 差异表达基因GO富集分析
对Campy 组与Mock 组肿瘤与癌旁组织三种比较组筛选出来的显著性差异基因进行GO 富集分析,以P<0.05 作为筛选标准。GO 富集分析主要包括三个部分,分子功能(molecular function,FM)、生物学过程(biological process,BP)以及细胞组分(cellular component,CC)。通过GO 富集分析结果可以看出,对比Campy 组肿瘤部位与癌旁部位,如图4-1 所示,差异基因中的上调基因在生物学过程模块中,主要富集在肽酶活性的调节、白细胞的迁移、伤口愈合、白细胞细胞-细胞黏附等,分子功能模块中,细胞因子活性、受体配体活性、细胞因子受体结合通路富集程度较高;下调基因在生物学过程中表达量显著性差异较大的基因主要富集在脂肪酸代谢通路,其余还有脂肪细胞分化、棕色脂肪细胞分化通路等。
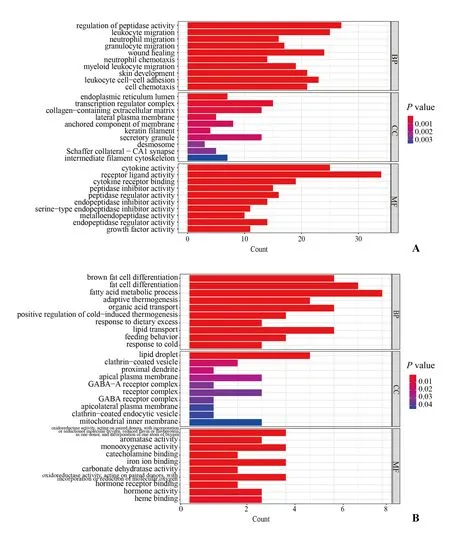
图4-1 Campy-tumor 组对比Campy-para 组差异基因GO富集分析Fig.4-1 GO enrichment of DEGs from Campy-tumor group vs.Campy-para group
可以看出,C.jejuni作用下的小鼠结直肠癌的发生、发展借助表面受体与细胞外基质的黏附因子特异性结合,进而诱导受体激活,并通过一系列的蛋白水解、跨膜运输等影响肿瘤细胞的生长、侵袭和转移。
对比Campy组与Mock组的肿瘤组织,如图4-2所示,上调基因中除了主要富集在膜、刷状缘相关的细胞组分通路外,在生物学过程中主要富集在阴离子转运、调节其他生物、抗菌肽介导的抗菌体液免疫反应、胆固醇平衡和粘膜固有免疫途径以及分子功能通路的脂质转运蛋白、血红素结合以及跨膜转运蛋白活性等;下调基因中主要富集在生物学过程中的多种免疫相关通路包括单核细胞、淋巴细胞、B 细胞、T 细胞等的增殖和活化通路调节,分子功能通路上主要富集到的通路是GTP酶、碳水化合物、细胞因子等多种蛋白质的结合和调节通路。
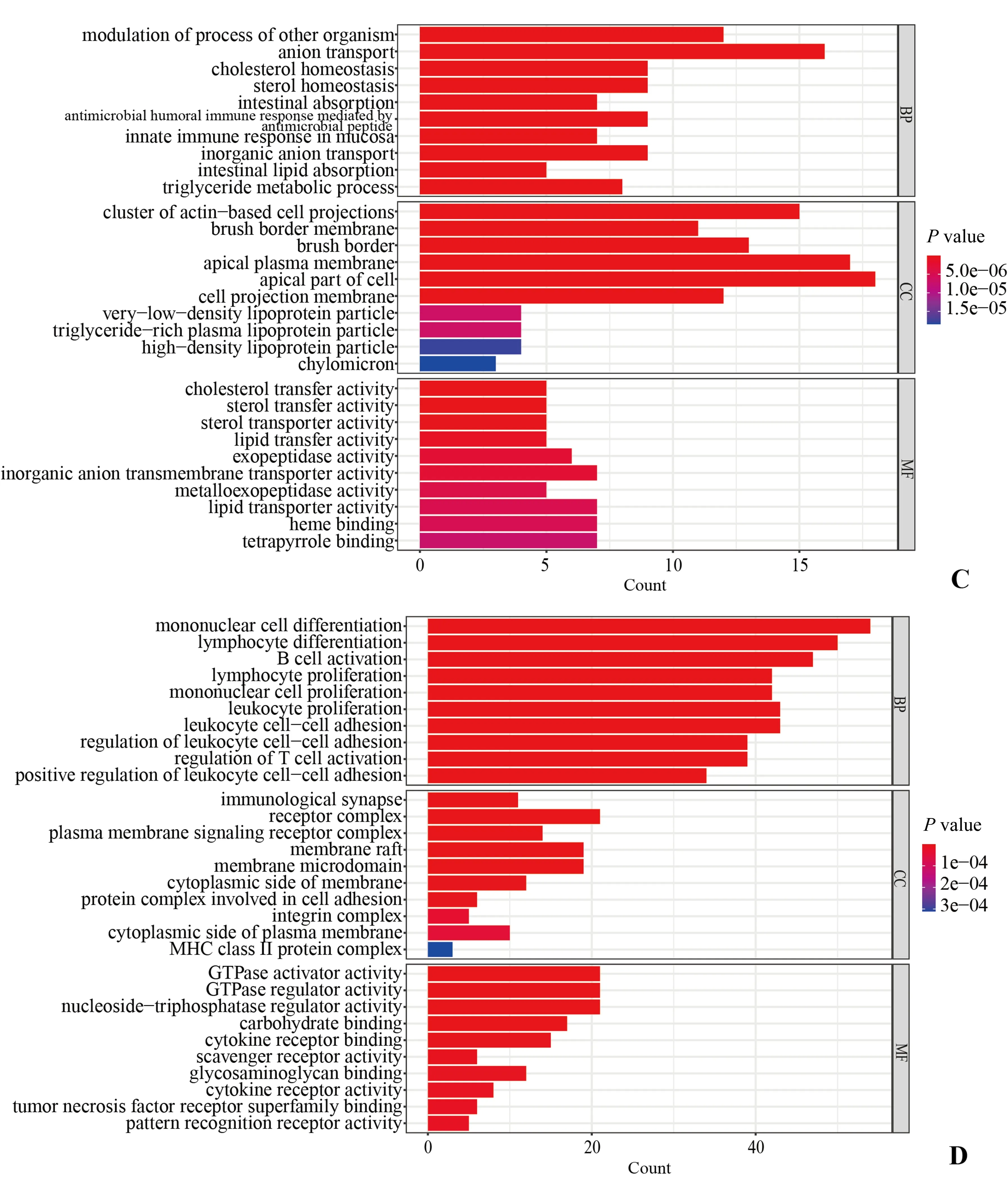
图4-2 Campy-tumor组对比Mock-tumor组差异基因GO富集分析Fig.4-2 GO enrichment of DEGs from Campy-tumor group vs.Mock-tumor group
对比Campy组和Mock组的癌旁部位,如图4-3所示,上调基因中显著性差异较大的主要富集在生物学过程中的其他生物膜破裂、抗菌肽介导的抗菌体液免疫反应、抗菌体液反应、防御细菌反应和粘膜固有免疫途径以及分子功能中的多种物质的跨膜转运活性,与Campy 与Mock 组的肿瘤部位相似;下调基因中基因表达差异较大的主要富集在生物学过程中的消化途径和分子功能的丝氨酸类肽酶活性和丝氨酸水解酶活性途径。

图4-3 Campy-para组对比Mock-para组差异基因GO富集分析Fig.4-3 GO enrichment of DEGs from Campy-para group vs.Mock-para group
2.5 KEGG通路分析
对三种对比组的差异基因进行KEGG 通路富集分析,判断基因富集的阈值为P<0.05。我们分别挑选了基因上调和下调的富集最显著的前10 条通路进行展示(其中Campy-tumor 组对比Campypara 组的下调基因只富集在6 条通路),如图5 所示。对于Campy-tumor组和Campy-para组,细胞因子和细胞因子受体相互作用通路是上调基因中差异最显著且富集最多的通路,其余还有IL-17 信号通路和Wnt信号通路等;富集最多显著性最高的下调基因通路是PPAR 信号通路,其次是神经活性配体-受体相互作用。对于Campy-tumor 组和Mocktumor组,上调基因中富集最多的是代谢途径,富集最显著的是脂肪的消化和吸收;下调基因中富集最多且最显著的是细胞黏附分子,其余还有细胞因子与细胞因子受体的互作、癌症中的通路和NF-κB信号通路等。关于Campy-para 组和Mock-para 组,上调基因富集最多的为代谢途径,其余还有癌症中的转录失调、金黄色葡萄球菌感染和NOD 样受体信号通路等;下调基因富集最多且最显著的是胰腺分泌,其余还有代谢途径、蛋白质的消化和吸收等。

图5 KEGG富集分析Fig.5 KEGG enrichment analysis
2.6 韦恩图法分析
为了明确C.jejuni诱导的结直肠癌(Campy-tumor 组)的基因表达特点,本研究设计了两个对照,分别为Campy-para 组和Mock-tumor 组。前述分析可知,Campy-tumor 与对照组Campy-para 相比,共发现394 个差异表达基因;而Campy-tumor 与另一个对照组Mock-tumor相比,共有501个差异表达基因;结果显示,有17 个差异表达基因(图6A 红蓝交汇处),在两次对照中均有出现,意味着它们在C.jejuni诱导的结直肠癌的发生发展可能起到重要作用。
两个对照组之间的比较,即Mock-tumor 与Mock-para 相比,得到1 008 个差异表达基因(黄色区域)。这些基因表达变化是Apc基因缺失导致的癌变本身即具有的,它们的表达变化不依赖于C.jejuni的诱导。另一方面,Campy-para 与Mock-para相比,得到316 个差异表达基因(绿色区域)。这些基因表达变化与C.jejuni感染有关,但与癌变这一结果不相关。因此,为了得到更加严谨的结果,我们从上述红蓝交汇处的17 个基因减去了与黄色区域交汇的1 个基因(红蓝黄交汇区)和与绿色区域交汇的2 个基因(红蓝绿交汇区),剩下14 个基因。这14 个基因表达的变化在C.jejuni诱导的结直肠癌的发生发展中可能起到了重要且独特的作用,因此我们称其为“核心差异表达基因”。其中,有5 个基因(Lipf、Gm1987、Ascl5、Saxo1和Plekhs1)表达上调,9 个基因(Lrp2、Phex、Serpina3c、Fabp4、Vwa3a、Tmem52、Lrrn4、Upk3和Crb2)表达下调(表2)。其中表达量差异超过30 倍的有:Lipf(1×106倍)、Gm1987(1×103或1×106倍,取决于不同的对照组)、Ascl5(约32倍)、Phex(约32倍)。
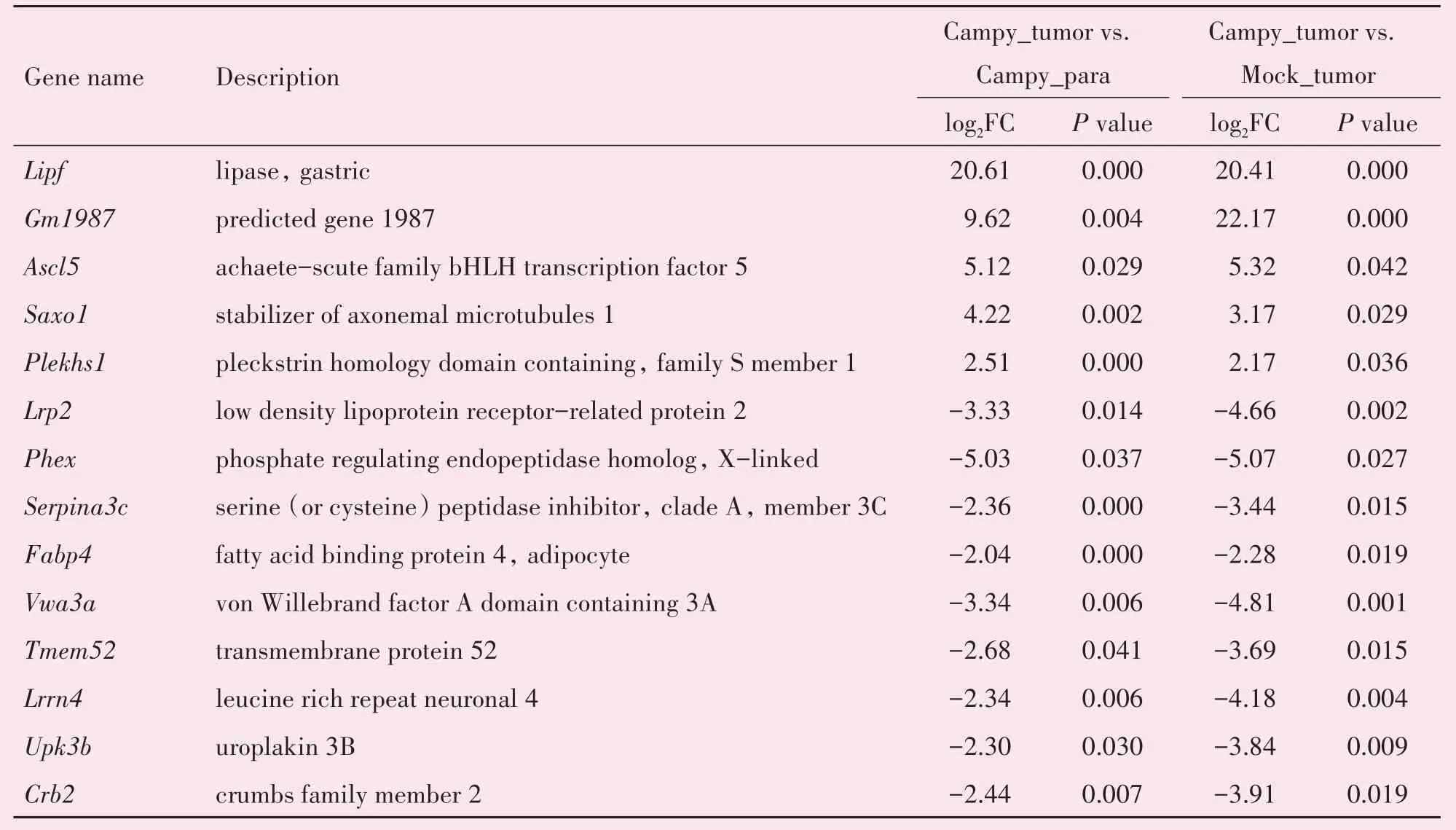
表2 14个核心差异表达基因Table 2 Fourteen core DEGs
上述14 个基因的组间表达量聚类图、GO 注释信息和KEGG 注释信息如图6(B-F)所示。可以看到,在GO 注释信息里的生物学过程模块里这些差异表达基因主要参与了细胞过程、生物调节、生物过程的调控、对刺激的反应、代谢通路等过程;在分子功能模块,主要参与了结合功能;在细胞组分模块,主要与细胞和膜相关。在KEGG 通路注释信息里,这些核心差异表达基因主要参与了代谢相关通路和癌症发生相关通路,代谢相关通路有:代谢途径、胆固醇代谢、脂肪消化吸收、脂肪细胞里的脂肪分解调节、甲状腺激素的合成和甘油酯类代谢通路,与癌症相关的通路包括了细胞因子-细胞因子受体互作、趋化因子信号通路、河马信号通路、NFκB信号通路、PPAR信号通路、刺猬信号通路。

图6 韦恩图法分析与14个核心DEGs的功能分析Fig.6 Venn diagram analysis and the resulting 14 core DEGs with functional analysis
2.7 qRT-PCR 对RNA-seq结果的验证
我们用qRT-PCR 对上述14 个差异表达基因进行验证。与Campy-para 相比,Campy-tumor 中Lipf、Gm1987、Ascl5、Saxo1、Plekhs1mRNA 表达上调,Lrp2、Phex、Serpina3c、Fabp4、Vwa3a、Tmem52、Lrrn4、Upk3b、Crb2表达下调,升降趋势与RNA-seq结果完全一致,其中有9 个基因(Gm1987、Saxo1、Plekhs1、Lrp2、Serpina3c、Fabp4、Tmem52、Lrrn4、Upk3b)的表达两组间具有显著性差异(P<0.05;图7),表明测序结果重复性较好,可信度较高。

图7 14个核心DEGs的qRT-PCR结果Fig.7 qRT-PCR results of 14 core DEGs
3 讨论
已知C.jejuni对宿主造成疾病主要是依赖运动、黏附、侵袭和产毒素这些毒力因子[13],其产生的细胞致死性膨胀毒素(cytolethal distending toxin,CDT)是目前已知的促进结直肠癌发生的关键因素[7],然而宿主基因是如何参与C.jejuni诱导癌症发生的,这一点仍不清楚。因此,本研究通过转录组学分析,以揭示在C.jejuni诱导的肿瘤中表达改变的宿主基因。
ApcMin/+小鼠是研究肠道肿瘤发生、发展过程的经典模型[14],通过给ApcMin/+小鼠灌胃C.jejuni81-176 株细菌诱导小鼠结直肠癌的发生,再用葡聚糖硫酸钠(dextran sulfate sodium,DSS)破坏肠上皮屏障加快建模。通过评估小鼠的成瘤情况,我们发现C.jejuni诱导下的小鼠肿瘤数量显著高于对照组,表明C.jejuni可以促进小鼠结直肠癌的发生,与前人的研究[7]相符。在已报道的动物模型中[7],观察到C.jejuni诱导的CRC 有两个先决条件:即宿主的Apc基因突变和有一个致炎环境(如DSS处理)。因此,检测到的基因表达变化可以被描述为当前CRC模型(即包含Apc-和DSS 前提)下,C.jejuni诱导的CRC基因表达变化。
通过较为严谨的对照,我们指出了14 个可能在C.jejuni诱导的结直肠癌发生发展中起到重要且独特作用的基因。经过进一步的qRT-PCR 验证,14 个基因中的9 个(Gm1987、Saxo1、Plekhs1、Lrp2、Serpina3c、Fabp4、Tmem52、Lrrn4、Upk3b)在结直肠肿瘤和癌旁部位的表达具有统计学上的显著性差异。其中关于表达上调的基因,Plekhs1在人类里也有同源基因,有研究表明它的过表达增强了蛋白激酶B 的磷酸化,继而增加致癌活性,促进癌细胞增殖、存活和侵袭,该基因能促进甲状腺癌的恶化[15],也可以作为膀胱癌[16]和胃癌[17]的基因标志物,但目前关于它的表达是如何被调控的以及和结直肠癌的关系尚不清楚。结直肠作为临近胃和膀胱的一个器官,C.jejuni很可能通过某些方式引起该基因的高表达,继而引发结直肠癌的发展。Saxo1编码微管稳定蛋白,对纤毛等有特异性,目前关于它的报道主要关注在小鼠精子活力方面,鉴于我们的实验动物是雌性小鼠,所以该基因的具体作用还有待探究。还有一个值得关注的基因是Gm1987,目前尚没有在Pubmed 数据库里发现有关该基因的数据,但它在我们的研究中提示参与了多条信号通路,包括有细胞因子-细胞因子信号通路、NF-κB信号通路以及趋化因子信号通路,这些都是癌症发生发展过程中极其重要的通路,所以该基因值得进一步探究。
关于我们筛选出的C.jejuni诱导结直肠癌有关的表达下调的6 个核心差异表达基因,只有2 个基因有被报道与CRC 有关。Fabp4是一种编码脂肪酸结合蛋白4 的基因,人类基因组上也有同源基因。有报道表明在CRC 患者的肿瘤细胞中FABP4蛋白表达降低,推测FABP4 参与了PPARγ 通路促进结直肠癌细胞的迁移和侵袭[18]。Lrrn4编码富含亮氨酸重复序列神经元4 蛋白,先前的一项研究发现,LRRN4 在正常结肠组织中的丰度较低,在CRC组织中的水平更低[19]。然而前不久的一项研究表明它在CRC 中无论是mRNA 还是蛋白水平的表达都较高,其通过RAS∕MAPK 信号通路调节肿瘤细胞的几种恶性表型[20]。这种矛盾的结果可能是由于方法或者样本数不同引起的。还有一篇文献报道了Lrrn4和我们筛选的另一个表达下调的基因Upk3b它们编码的蛋白LRRN4 和uroplakin 相较于原代间皮细胞在间皮瘤内表达下调或缺失[21],但与CRC 相关的研究目前没有任何报道。Lrp2编码低密度脂蛋白相关蛋白2 或维生素D 相关转运蛋白megalin,它在结肠等多个上皮细胞系中表达[22],目前还没有研究调查Lrp2与CRC 之间的可能关联,但与Lrp2相关的基因Cubn曾在一项荟萃分析里报道与CRC 存在一定的关系[23]。Serpina3c是小鼠中人类SERPINA4基因的一个分支,是调节脂肪生成过程中信号网络的关键因素,它可以通过Wnt∕βcatenin-PPARγ途径参与脂肪分化的调节,在Serpina3c-/-小鼠中引起脂肪细胞分化受损[24]。虽然目前没有报道它与癌症的直接关系,但脂肪代谢的紊乱往往会促进癌症的发展。关于Tmem52基因,有少量的文献介绍其在肺腺癌组织中表达上升[25]。总得来说,我们推测可能是这些表达下调的基因所编码的产物没有为肿瘤提供竞争优势,所以在肿瘤部位的表达会受抑制。有趣的是,在我们的研究中筛选出来的核心差异表达基因,大多数目前还没有被报道与CRC 之间的关系,这值得将来进一步的探索。
本实验通过对筛选出来的差异基因进行GO富集、KEGG 通路富集分析,Campy组与Mock组相比,差异基因参与了机体许多重要的生物学过程:生物膜破裂、蛋白质水解、信号转导通路以及与细菌引起的免疫反应相关的通路:抗菌肽介导的抗菌体液免疫反应、抗菌体液反应、防御革兰氏阳∕阴性细菌反应和粘膜固有免疫途径。之前的研究表明,通过对C.jejuni81-176感染的无菌ApcMin/+小鼠的远端结肠组织进行转录组测序,C.jejuni感染的小鼠中有两条致癌途径富集——神经活性配体-受体相互作用和细胞因子-细胞因子受体相互作用[7],在我们的Campy 组与Mock 组对比以及Campy 组的肿瘤与癌旁组织对比中也有这两条通路的富集。此外,代谢通路在Campy 组对比Mock 组中富集最显著,提示着C.jejuni感染下代谢反应相当活跃。综上,差异基因参与了多条重要的生物学通路,提示结直肠癌的发生发展通过代谢、蛋白转运等相关通路进行。需要指出的是,本研究campy-tumor-2 号小鼠的基因表达情况与该组其它小鼠差异明显,然而该鼠的肿瘤数量(4 个)与该组平均水平(4 个)相当。我们认为此小鼠组学数据的不同是个体差异性所致,我们将在后续研究中扩大样本量来平衡个体差异问题。
综上所述,本研究通过较为严谨的对照,指出了9 个可能在C.jejuni诱导的结直肠癌的发生发展中起到重要且独特作用的基因:Gm1987,Plekhs1,Saxo1,Lrp2,Serpina3c,Fabp4,Tmem52,Lrrn4,Upk3b。为后续研究这些基因在C.jejuni诱导的结直肠癌中的作用提供了新的方向和思路。需要指出的是,关于这“9 个宿主基因”,我们目前得出的结论是“可能”参与肿瘤的发生发展,我们将在后续的研究中扩大样本量,并对上述基因进行进一步的功能验证。
猜你喜欢
中国典型病例大全(2022年11期)2022-05-13
中国典型病例大全(2022年7期)2022-04-22
中国典型病例大全(2022年7期)2022-04-22
现代仪器与医疗(2021年4期)2021-11-05
现代仪器与医疗(2021年1期)2021-06-09
科学导报(2021年29期)2021-06-03
科海故事博览·下旬刊(2019年6期)2019-04-16
中国中药杂志(2017年15期)2017-08-30
中国中药杂志(2017年15期)2017-08-30
江苏农业科学(2017年5期)2017-04-15
