
铕掺杂对Co3O4活化过一硫酸盐降解亚甲基蓝性能的增强
2022-08-09 03:49廖小刚沈海丽
无机化学学报 2022年8期
夏 强 廖小刚 沈海丽 郑 林 李 纲 沈 俊
(重庆理工大学化学化工学院,重庆 400054)
0 引言
高级氧化技术(AOPs)是深度处理有机废水的有效手段,其对有机污染物的快速降解主要依赖于体系中生成的各种活性氧物种。近来,基于过一硫酸盐(PMS)的AOPs引起了人们的极大关注。该技术以水溶性的PMS为氧化剂,通过一定的方式将其活化后,即可在溶液中产生SO4-·、·OH、·O2-和1O2等活性氧物种,借此来实现对有机污染物的净化[1-2]。与以往的AOPs相比,该技术以在反应体系中产生比·OH氧化还原电势更高的自由基SO4-·为标志和特点。
PMS的活化方式包括能量活化法(如光、热、超声波和微波活化等)和催化活化法两类。催化活化法特别是基于过渡金属氧化物等的非均相催化活化法,因具有无需外加能量、操作简单且催化剂可循环使用等优点而备受青睐。然而,与均相催化剂和PMS间的高效单相反应相比,非均相催化剂依靠其表面配位金属离子来活化PMS的异相催化方式则作用有限,其对PMS的活化性能有待进一步提高。为提升非均相催化剂活化PMS的效果,目前主要形成了2种解决思路。一是从催化材料的成分和微观结构层面进行设计或改良,以此增强催化剂的活性,如掺杂异种离子[3-5]、构筑复合材料[6-8]或对催化剂的表面改性[9]等;二是对催化剂活化PMS的化学反应过程进行强化,通过辅助手段加快PMS在催化剂作用下的活化反应速率,如升高反应温度[7]、电化学强化[10]、超声辅助[11-12]或添加助催化剂[13-16]等。
过渡金属离子活化PMS的过程主要包含2步,即首先由低价过渡金属离子与PMS反应,生成氧化活性物种(如SO4-·等)和高价离子;生成的高价离子继续与剩余的PMS反应,其在被还原为低价离子的同时生成低氧化活性的中间体(如SO5-·等)。文献报道,向催化剂/PMS体系中加入羟胺[13]、原儿茶酸[14]或天然多酚[15]等还原性物质可以直接对高价金属离子进行还原;加入有机螯合剂[16]如L-抗坏血酸、N,N-二(羧甲基)-β-氨基丙酸等则可使高价金属离子更易于与HSO5-反应并被还原为低价态。这些方法均被证明可用于增强PMS的催化活化性能,其原理在于通过加速金属离子的高/低价态的循环,使失电子后低活性的高价金属离子能及时被还原为高活性的低价态,以此加快催化剂对PMS的持续活化。这类加速金属离子的高/低价态循环的手段是目前助催化剂法中普遍采用的研究思路,但值得注意的是,诸如天然多酚这些有机物的引入不仅会增大水体的总有机碳(TOC)含量,还可能造成二次污染,并且所加入的还原性物质还会与溶液中待降解的有机污染物竞争PMS,因此它们的添加量存在优化区间。而根据PMS活化机制分析,除加速活化过程中金属离子的高/低价态循环外,通过一定手段降低低价过渡金属离子与PMS反应难度同样可以达到增强催化剂对PMS活化效果的目的。如Xu等[17]的研究发现,向Co2+/PMS高级氧化体系中加入Ca2+、Ba2+等非氧化还原性金属离子(路易斯酸)后,体系对有机物的降解能力得到显著增强,并证明这是由于路易斯酸的吸电子效应增大了PMS的极性,使得其更易于被Co2+活化。亦即路易斯酸也可作为助催化剂增强过渡金属离子对PMS活化能力。
我们将路易斯酸强化PMS活化的方式应用于非均相催化剂Co3O4的设计与制备上,以期实现Co3O4催化活性的进一步增强。即以钴盐和铕盐为原料,采用草酸盐沉淀法先制备出草酸钴/铕前驱体,随后通过热分解该前驱体将路易斯酸性位Eu3+直接引入到催化剂Co3O4之中。以模拟染料废水亚甲基蓝(MB)溶液为降解模型,通过研究Eu的添加量与催化剂活化PMS降解MB的性能关系,得到优化的Co/Eu添加比例(nCo/nEu),最终获得具有高催化活性的Co/Eu双金属复合氧化物多孔材料。实验结果证实,Eu3+具有的强路易斯酸性特点成功增强了催化剂主成分Co3O4对PMS的活化能力,并极大地提升了其催化PMS降解MB的性能,这为其它非均相催化剂活化PMS性能的增强提供了新的思路和有益借鉴。
1 实验部分
1.1 实验原料
实验用到的试剂药品包括MB、过一硫酸氢钾、亚硝酸钠、六水合硝酸钴、六水合硝酸铕、一水合草酸铵、碘化钾、无水硫酸钠、氯化钠、草酸钠、碳酸氢钠、无水乙醇(EtOH)和叔丁醇(TBA),均为分析纯,由成都市科隆化学品有限公司提供;实验用水为去离子水,由GWA-UN2型超纯水器(北京普析通用仪器有限公司)制得。
1.2 实验方法
1.2.1 催化剂的制备
采用草酸盐热解法制备不同nCo/nEu的催化剂。具体步骤如下:在室温下,按一定比例(nCo/nEu=19、9、3、1、1/3)依次准确称取 Co(NO3)2·6H2O、Eu(NO3)3·6H2O共10 mmol,溶于100 mL去离子水中,记为溶液A;按照沉淀反应(式1、2)的化学计量比计算出将Co、Eu离子完全沉淀所需草酸根离子的物质的量n,然后准确称取1.05n的(NH4)2C2O4·H2O溶于105 mL去离子水中,记为溶液B;将溶液A、B预热至60℃,然后在强烈磁力搅拌下将溶液A逐滴加入至溶液B中,滴加完毕后继续搅拌2 h以使反应充分;真空抽滤,收集所得沉淀,并用去离子水和无水乙醇反复清洗,置于50℃干燥箱内干燥过夜;最后将前驱体放入马弗炉中,以2℃·min-1的升温速率升温至400℃并焙烧2 h,冷却后收集待用。为方便区分,将复合催化材料命名为CoaEub,nCo/nEu=a/b;另外,实验还单独以Co(NO3)2·6H2O或Eu(NO3)3·6H2O配制了溶液A,其他制备过程同上,所得样品被命名为Co3O4或 Eu2O3。

1.2.2 分析表征
样品的晶相结构和相纯度测试在XRD-7000型X射线衍射仪(XRD)上完成,CuKα辐射,X射线波长为0.154 06 nm,加速电压和电流分别为40 kV和30 mA,扫描范围2θ=10°~80°,扫描速率2(°)·min-1,扫描步长0.02°;样品的形貌通过日立Regulus8100型扫描电子显微镜(SEM)进行观察,加速电压为5 kV;样品的N2吸附-脱附测试在Micrometritics ASAP2020氮气吸附仪上完成,工作温度为-195.8℃;自由基捕获实验在JES FA200型电子自旋(顺磁)共振波谱仪(ESR/EPR)上完成,以 5,5-二甲基-1-吡咯啉-N-氧化物(DMPO)为 SO4-·、·OH 或·O2-捕获剂,4-氧-2,2,6,6-四甲基哌啶(TEMP)为1O2捕获剂;MB的浓度采用日本岛津UV-2700i型紫外可见分光光度计进行测定。
1.2.3 PMS浓度测定
PMS溶液浓度采用碘化钾分光光度法进行测定[18-20]。取2.5 mL 1.66 g·L-1的KI溶液(溶有NaHCO3用以防止 I-被 O2氧化,NaHCO3浓度 0.6 g·L-1)与 0.5 mL PMS溶液混合,然后于紫外可见光分光光度计上在波长352 nm处测定其吸光度,并根据标准曲线确定PMS浓度。实验中PMS初始浓度为0.6 mmol·L-1,加入催化剂后立即计时,并于给定时间点取样,所取样品中的催化剂颗粒经由0.22 μm混合纤维素滤膜滤去。
1.2.4 MB的催化降解实验
催化剂活化PMS降解MB的实验步骤如下:首先将催化剂加入至MB溶液中,搅拌混合50 min以达到吸附平衡;然后加入一定量的PMS启动反应,在设定时间点取样并测定MB溶液浓度,获取MB浓度随反应时间的变化关系,以此分析Co/Eu复合催化剂活化PMS降解MB的性能。MB溶液的初始浓度为10 mg·L-1,体积为500 mL;为避免反应温度对PMS活性的影响,所有降解实验均在25℃恒温水浴下进行(考察温度影响时除外);每次取样体积为3.5 mL,所取样品先用0.22 μm尼龙滤膜过滤,然后立即注入0.15 mL浓度3 mol·L-1的NaNO2猝灭反应,随后在500~700 nm波长下测定其最大吸光度值。MB降解率(η)通过下式进行计算:

其中c0为达到吸附平衡时溶液中的MB浓度;ct为反应进行到tmin时溶液中的MB浓度。
2 结果与讨论
2.1 材料表征分析
2.1.1 XRD分析
图1为按不同nCo/nEu制备的催化剂的XRD图。由图可见,纯铕氧化物没有明显的衍射峰,仅在2θ=30°处出现“馒头峰”,表明以草酸盐热解法制得的铕氧化物不具备有序的晶体结构,其原子混乱排布,属于非晶态材料,为方便区分,将其命名为Eu2O3;纯钴氧化物衍射峰明显,峰型尖锐,半峰宽窄,与立方晶系Co3O4(PDF No.42-1467)相匹配,表明以草酸盐热解法制得的钴氧化物是结晶度良好的Co3O4;复合金属氧化物样品的各主要衍射峰均与立方晶系Co3O4(PDF No.42-1467)相匹配,表明其中含有Co3O4晶体,而Eu2O3则以无定型态方式存在。随着Eu相对含量的增加,样品的衍射峰强度逐渐减小,表明Eu的加入降低了复合材料的结晶度。
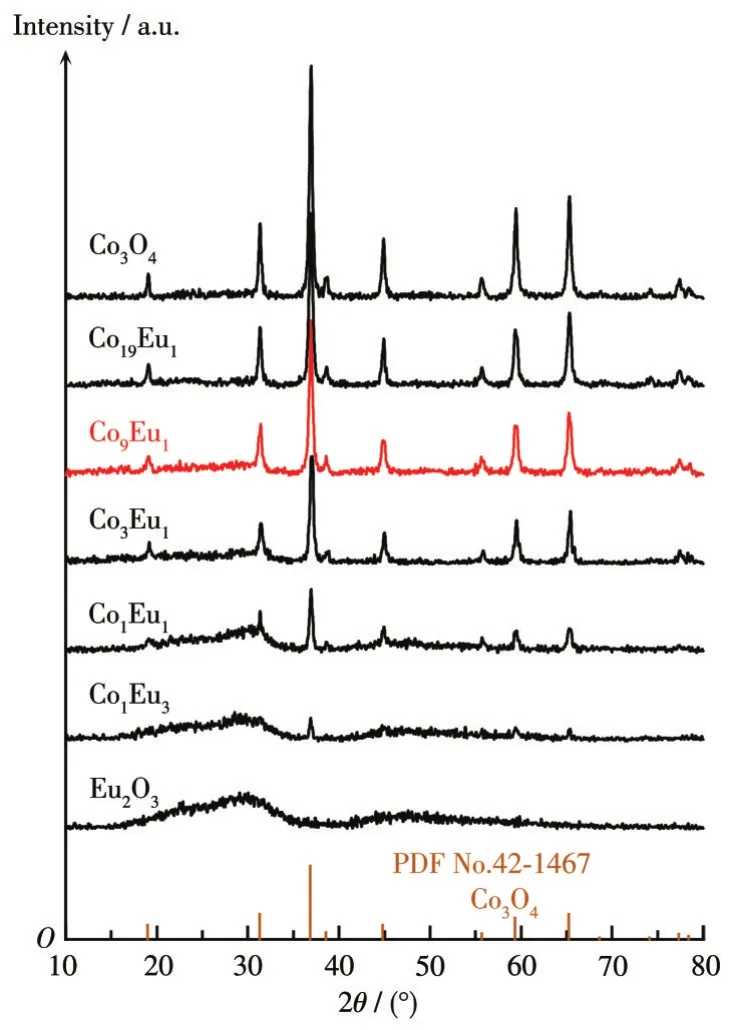
图1 不同Eu含量催化剂的XRD图Fig.1 XRD patterns of the catalysts with various Eu contents
2.1.2 SEM分析
不同Eu含量催化剂的SEM照片如图2所示。其中Co3O4呈米粒状,长约10 μm,宽约2 μm,其表面粗糙,因草酸盐前驱体热解而产生的大量裂隙清晰可见;随着Eu含量的增加,样品颗粒的长径比逐渐减小,并由米粒状最终变为块状,同时样品表面的裂隙逐渐缩小,表明Eu的掺入引起了材料微观形貌的变化。从图2g中则可观察到Eu2O3颗粒具有光滑表面,无明显孔隙结构;图2h~2k为Co9Eu1的能谱结果,样品中主要检测到O、Co、Eu三种元素,且分布均匀,证实以草酸盐热解法可以制备出均匀复合的钴铕双金属氧化物材料。

图2 不同Eu含量催化剂的SEM图(a~g)及Co9Eu1的能谱结果(h~k)Fig.2 SEM images of the catalysts with various Eu contents(a-g)and energy spectrum results of Co9Eu1(h-k)
2.1.3 比表面积和孔结构分析
图3为不同Eu含量催化剂的N2吸附-脱附等温线及孔径分布曲线结果。由图可见,Co3O4、Eu2O3以及不同nCo/nEu样品的N2吸附-脱附等温线相似,均为第Ⅳ类Langmuir吸附-脱附等温线线型。除Co3O4和Co3Eu1的吸附支与脱附支基本重合外,其余样品的吸附支与脱附支均不重合,形成了明显的H3型迟滞环,这是由毛细管凝集引起的,表明这些样品具有良好的介孔结构,且其为层状粒子堆叠而成的狭缝型孔道。孔径分布曲线结果显示,所有样品的孔径均集中在2~50 nm之间,证实其均为介孔材料。采用Barrett-Joyner-Halenda(BJH)方法由吸附支计算出的样品比表面积及孔结构参数列于表1。由表中数据可知,少量Eu的掺入有助于增大材料的比表面积和改善材料的孔结构,但随着复合材料中Eu含量的增加,其比表面积和孔容值均逐渐减少。
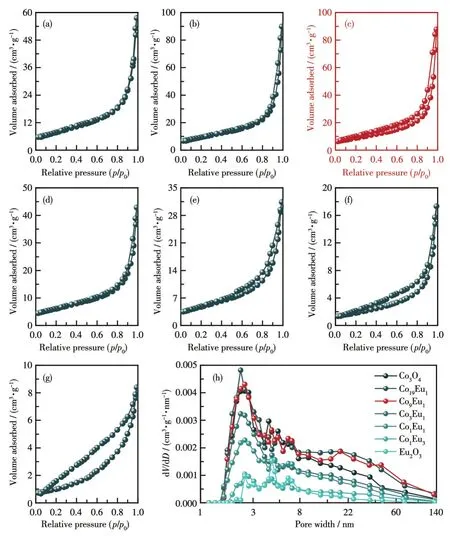
图3 不同Eu含量催化剂的N2吸附-脱附等温线(a~g)及孔径分布曲线(h)Fig.3 N2adsorption-desorption isotherms of the catalysts with various Eu contents(a-g)and pore size distribution curves(h)
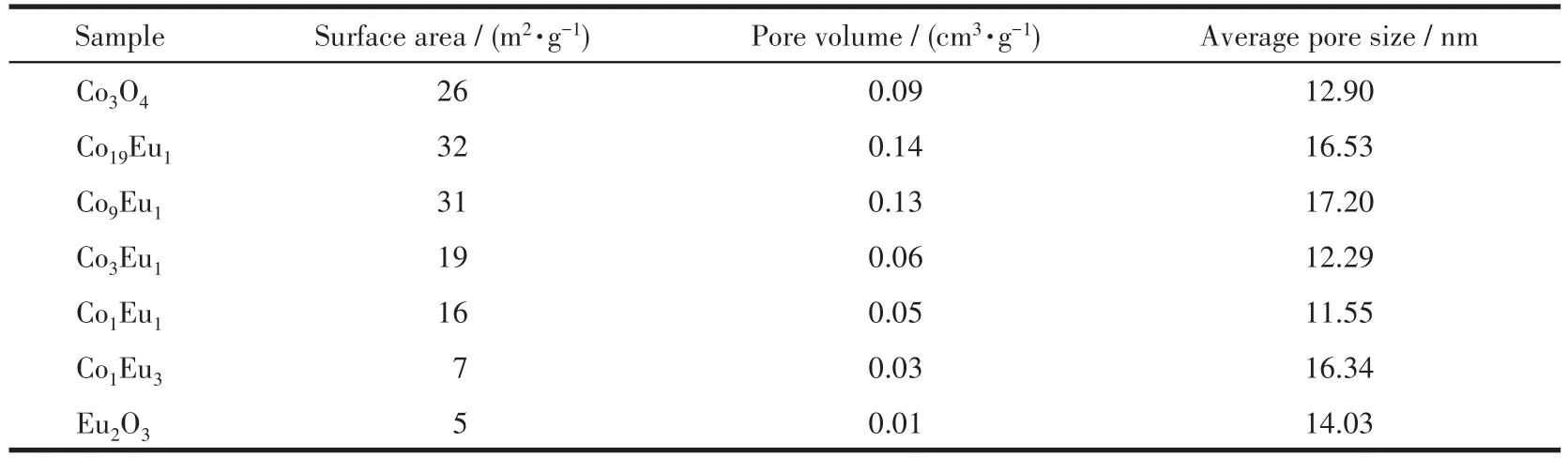
表1 不同Eu含量催化剂的比表面积及孔结构参数Table 1 Specific surface area and pore structure parameter of catalysts with various Eu contents
2.2 不同nCo/nEu催化剂的性能对比
2.2.1 PMS分解效果对比
为了对比不同nCo/nEu的催化剂在活化PMS降解MB过程中的活性,分别设计了PMS分解实验和MB催化降解实验,结果如图4所示。实验中催化剂用量均为 0.10 g·L-1,PMS 浓度均为 0.6 mmol·L-1。从PMS分解实验结果来看(图4a,c0'、ct'分别为PMS的初始浓度及分解t时刻时的浓度),Co3O4和Eu2O3对PMS的分解效果明显较差,在其作用下PMS的分解反应速率常数分别为0.005 1和0.005 8 min-1。相比之下,复合材料对PMS的分解效果均显著优于单金属氧化物,并且随Eu含量的增加先升高而后降低,其中nCo/nEu=3的复合金属氧化物材料对PMS分解效果最佳,其催化分解PMS的反应速率常数达0.059 2 min-1。PMS在不同催化剂作用下分解速率快慢依次 为 Co3Eu1>Co1Eu1>Co9Eu1>Co1Eu3>Co19Eu1>Eu2O3≈Co3O4。
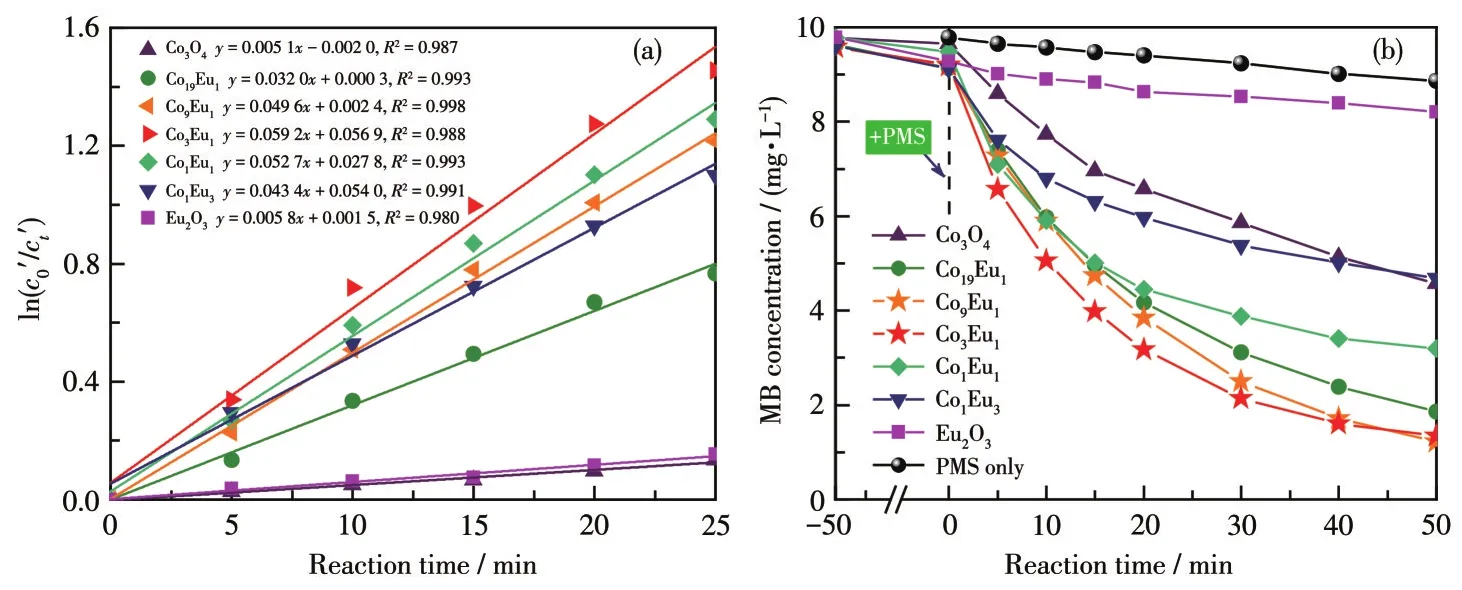
图4 不同Eu含量催化剂对PMS的催化分解性能(a)及相应PMS活化体系对MB降解效果(b)Fig.4 Decomposition performance of PMS catalyzed by the catalysts with various Eu contents(a)and MB degradation effect in the corresponding catalyst/PMS reaction system(b)
2.2.2 MB降解性能对比
从相应的催化剂/PMS反应体系对MB的降解实验结果可知(图4b),Co/Eu双金属氧化物复合材料活化PMS降解MB的效果明显优于单金属氧化物,其中以Co3Eu1/PMS和Co9Eu1/PMS体系对MB的降解效果最优。值得注意的是,不同材料催化PMS降解MB的性能与其对PMS的活化分解作用之间并不呈正相关关系。例如纯Co3O4和Eu2O3催化分解PMS的速率近乎相等(图4a),但MB在Co3O4/PMS体系中的降解率显著高于Eu2O3/PMS体系(图4b);而在不同nCo/nEu下制备的5种复合材料中,Co19Eu1催化分解PMS的速率最低,但其活化PMS降解MB的效果却明显优于Co1Eu1及Co1Eu3,这是由于PMS在Co3O4和Eu2O3的催化作用下分解路径不同:Co3O4中Co2+位点是公认的催化活性物质,在其作用下PMS以单电子转移方式分解并产生各种高氧化活性的中间体,该分解方式对MB的氧化降解过程起到重要作用;而Eu2O3中Eu3+作为强路易斯酸,主要诱导PMS以双电子转移的方式分解并产生低氧化活性的O2[17],该分解方式对促进MB的氧化降解几乎毫无意义。
上述实验结果表明,形成Eu2O3/Co3O4双金属复合氧化物材料可以提升PMS氧化降解MB的能力,这一方面得益于Co、Eu元素间的协同作用,即Eu3+的强路易斯酸和缺电子特性增强了对吸附在催化剂表面PMS的极化程度,使得PMS更易于接受来自Co3O4中Co2+位点的电子而被活化;另一方面,前文已证实少量Eu的掺入可提高材料的比表面积,这也有助于增加用于参加反应的活性位点数量。同时不难看出,Eu掺入量有一定的合适范围,以nEu/nCo<1为宜,即形成以Co3O4为主催化成分的复合催化剂体系更有利于对MB的催化降解。对本文而言,Co3Eu1和Co9Eu1表现出相当的活化PMS降解MB的性能,但考虑到稀土铕盐的价格更为昂贵以及路易斯酸性位点Eu3+含量过多时引起的PMS无效分解,我们确定出nEu/nCo更小的Co9Eu1更适合作为催化剂用于染料分子的降解,故后续实验均选择该样品作为研究对象。
2.3 MB催化降解实验
2.3.1 催化剂用量、PMS浓度及反应温度的影响
图5为不同工艺参数对Co9Eu1催化PMS降解MB的影响实验结果。图5a是PMS浓度固定为0.6 mmol·L-1时不同催化剂用量对MB的降解影响结果。当催化剂用量从 0.05 g·L-1增加至 0.20 g·L-1,MB的降解率分别为53.22%、86.66%、92.87%和94.07%,说明增加反应体系中的催化剂用量有助于提高其对MB的催化降解效果。这是因为催化剂用量的增加可以为反应提供更多的催化活性位点,进而加速对体系中PMS的活化速率以及体系对MB的氧化降解速率。
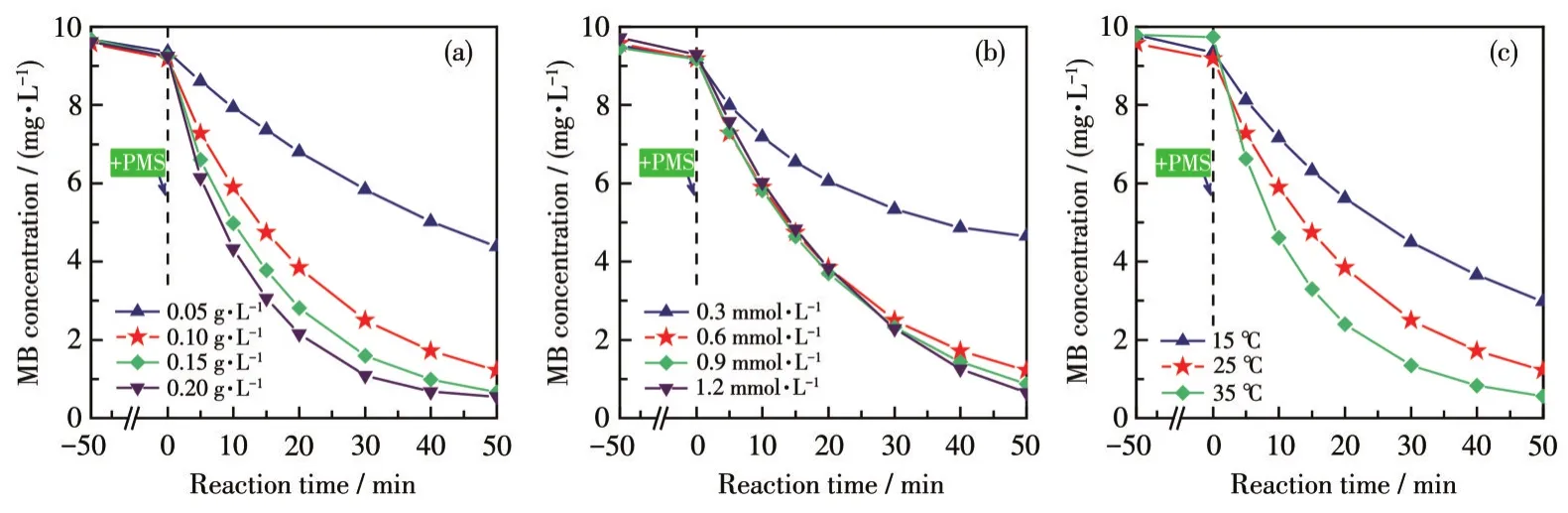
图5 催化剂用量(a)、PMS浓度(b)和反应温度(c)对Co9Eu1/PMS反应体系降解MB性能的影响Fig.5 Effect of catalyst dosage(a),PMS concentration(b),and reaction temperature(c)on MB degradation rate in Co9Eu1/PMS reaction system
图5b对比了催化剂用量为0.10 g·L-1时不同PMS浓度对MB降解效果的影响。当PMS浓度由0.3 mmol·L-1增加至1.2 mmol·L-1时,MB降解率依次为49.27%、86.66%、90.46%和92.97%,即MB降解率与PMS浓度整体上呈正相关关系。但不难看出当PMS浓度达到 0.6 mmol·L-1后,继续增大 PMS 浓度对MB的降解过程促进作用不大,说明当催化剂用量为0.10 g·L-1时体系的饱和PMS浓度为0.6 mmol·L-1。当 PMS 浓度大于 0.6 mmol·L-1时,受制于反应体系中有限的催化剂表面活性位点数量,剩余的PMS难以参与到对MB的降解反应中,因此对MB的降解提升不大。
图5c是反应温度对MB降解率的影响的实验结果。实验时催化剂用量和PMS浓度分别为0.10 g·L-1和0.6 mmol·L-1。由图可见,MB的降解速率随温度上升呈现出明显的加快趋势,在15、25和35℃下其最终降解率分别为68.14%、86.66%和94.19%。MB降解率的提升一方面源于温度升高引起的化学反应速率加快,另一方面也与PMS受到了温度的热活化相关。
2.3.2 催化剂循环使用性能
采用“过滤→洗涤→干燥”的方式对使用后的催化剂进行回收,并对其循环使用性能进行测试。实验时催化剂用量和PMS浓度分别为0.10 g·L-1和0.6 mmol·L-1。图6a显示,在连续的4次循环实验中,MB降解率分别为86.66%、71.99%、80.10%和78.08%,表明复合材料Co9Eu1具有良好的循环使用性能。同时,还通过XRD和SEM表征手段分析了4次循环使用后Co9Eu1催化剂的晶相结构及微观形貌的变化。由图6c可知,循环使用前后催化剂的XRD图几乎没有任何差异,表明循环使用过程中Co9Eu1的晶相结构没有发生明显变化。图6d、6e显示,4次循环使用后Co9Eu1的微观形貌与使用前(图2c)基本一致,证明Co9Eu1的微观结构在循环使用过程中亦未受到破坏。以上测试结果证实,利用本研究的方法制得的复合材料Co9Eu1具有稳定的机械性以及物理化学性质,在催化活化PMS过程中性能稳定,且可多次重复使用,是一种极具应用前景的PMS活化候选材料。
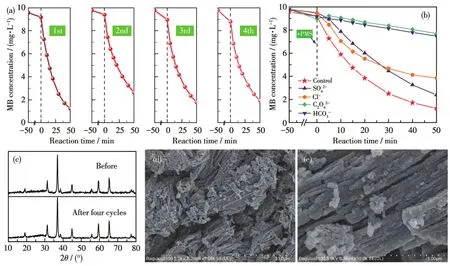
图6 催化剂的循环使用性能(a)、无机阴离子对MB降解率的影响(b)以及4次循环使用后催化剂的XRD图(c)和SEM图(d、e)Fig.6 Reuse-ability of the catalyst(a),effect of inorganic anions on MB degradation rate(b),and XRD patterns(c)as well as SEM images(d,e)of the catalyst after four cycles
2.3.3 常见无机阴离子的影响
对于高级氧化体系而言,实际废水中与有机污染物共存的各种无机阴离子对水处理效果的影响不容忽视。我们通过加入相应无机钠盐以探讨不同无机阴离子对Co9Eu1催化PMS降解MB过程的影响。实验时催化剂用量和PMS浓度分别为0.10 g·L-1和0.6 mmol·L-1,添加的4种无机钠盐浓度均为20 mmol·L-1,相应结果示于图6b中。由图可知,在加入SO42-、Cl-、C2O42-或HCO3-后,MB降解率分别下降为74.87%、59.37%、14.26%或18.53%,说明C2O42-和HCO3-对MB的降解具有明显抑制作用。可能的原因:(1)C2O42-具有较强的配位能力,其可能与催化剂表面金属离子发生配位,占据一定的催化活性点位;(2)C2O42-是典型的还原性离子,可与PMS活化产生的活性物反应或直接与PMS发生氧化还原反应,即与MB分子竞争氧化剂;(3)Na2C2O4和NaHCO3的引入会改变溶液pH,导致溶液呈弱碱性进而影响Co9Eu1/PMS体系对MB的降解能力;(4)HCO3-是典型的SO4-·和·OH猝灭剂[21],其存在大大阻碍了SO4-·和·OH对MB的降解。另外,Cl-的抑制作用主要由于其与活性物种反应生成了较低氧化能力的·Cl或ClO-,SO42-的抑制作用则可能源自其大量存在而带来的SO4-·的氧化还原电位的降低。
2.3.4 反应机理分析
采用自由基猝灭实验对Co9Eu1/PMS体系中的SO4-·和·OH进行鉴别,选择TBA作为·OH猝灭剂,EtOH作为SO4-·和·OH共同猝灭剂[22-25]。反应体系中催化剂用量和PMS浓度分别为0.10 g·L-1和0.6 mmol·L-1,猝灭剂用量为 200 mmol·L-1。图7a显示,当反应体系中加入TBA或EtOH后,MB的降解率分别从对照组的86.66%下降至79.54%或38.64%,表明·OH和SO4-·均参与了MB的降解反应,但SO4-·对MB的降解起主要作用。
进一步采用EPR测试技术对Co9Eu1/PMS反应体系中存在的活性含氧中间体进行直接检测,实验时催化剂用量为 0.10 g·L-1,PMS 浓度为 0.6 mmol·L-1。由图7b~7d可知,在反应体系中检测到DMPOSO4-·、DMPO-·OH、DMPO-·O2-和 TEMP-1O2四种加合物的特征信号峰,并且随反应时间的累积所有信号峰强度均逐渐增强,证明PMS在催化剂Co9Eu1的活化作用下发生了分解,并同时生成了SO4-·、·OH和·O2-三种自由基型活性物种以及1O2非自由基型活性物种。

图7 Co9Eu1/PMS反应体系中活性氧物种鉴定结果:猝灭实验(a);EPR谱图(b~d)Fig.7 Identification results of reactive oxygen species in Co9Eu1/PMS reaction system:quenching experiments(a);EPR spectra(b-d)
基于反应体系中活性物种的鉴定实验结果,提出如下的Co9Eu1活化PMS降解MB溶液的反应机理:
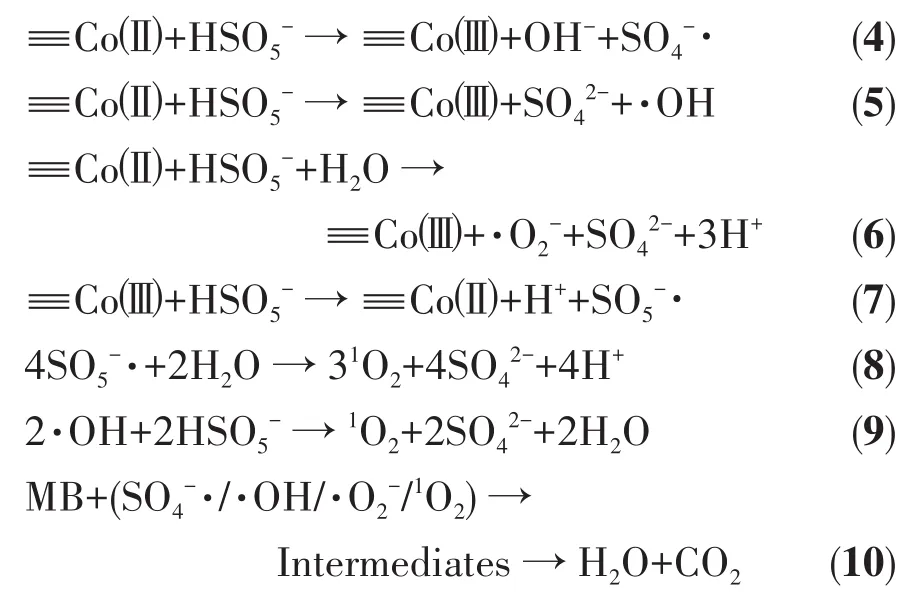
3 结论
采用草酸盐热解法制得了系列nCo/nEu不同的双金属复合氧化物多孔材料,并对其催化PMS降解MB溶液的性能进行评价。结果表明,稀土金属元素Eu的掺入可显著提高Co3O4对PMS的活化能力,这是Eu3+的缺电子特性可将吸附于催化剂表面的PMS极化,从而使其更易被Co3O4活化的结果。但过多Eu的掺入会导致PMS的无效分解,进而降低其活化PMS降解MB的性能。按nCo/nEu=9制得的催化材料(Co9Eu1)最具经济性和实用性,是一种极具潜力的非均相PMS活化材料。
在25℃反应条件下,当催化剂投加量和PMS浓度分别为 0.10 g·L-1和 0.6 mmol·L-1时,Co9Eu1/PMS高级氧化体系在50 min内对MB的降解率可达86.66%。催化剂Co9Eu1具有良好的稳定性,经历4次循环使用后,其对MB的降解率仍可达78.08%。Co9Eu1/PMS高级氧化体系对无机阴离子敏感,SO42-、Cl-、C2O42-以及HCO3-的存在均会抑制其对MB的氧化降解,其中C2O42-和HCO3-的抑制作用最为显著。自由基猝灭实验和EPR测试证明Co9Eu1/PMS体系中同时存在SO4-·、·OH 和·O2-三种自由基型活性物种与1O2非自由基型活性物种,且SO4-·是主要活性物种。
猜你喜欢
分子催化(2022年1期)2022-11-02
材料与冶金学报(2022年2期)2022-08-10
当代陕西(2022年5期)2022-04-19
建材发展导向(2021年16期)2021-10-12
科学与财富(2021年33期)2021-05-10
现代装饰(2021年1期)2021-03-29
智富时代(2018年3期)2018-06-11
智富时代(2018年3期)2018-06-11
中学生数理化·高一版(2016年7期)2016-12-07
试题与研究·中考化学(2016年1期)2016-09-30
