
VB2n-(n=8~12)团簇几何结构、电子及热力学特性的理论研究
2022-08-09 03:49李成刚崔颍琦邵琴琴申梓刚任保增
无机化学学报 2022年8期
李成刚 崔颍琦 田 浩 张 洁 邵琴琴 申梓刚 任保增
(1郑州师范学院物理与电子工程学院,郑州 450044)(2郑州大学化工与能源学院,郑州 450001)
0 引言
随着实验精度的不断提升和计算机技术的迅速发展,具有新颖几何结构、独特物理和化学特性的团簇引起了研究者广泛关注[1-3]。B原子的特殊电子结构,使其易与其它原子成键,在构建多功能纳米材料及调控其性能时表现出独特的优势,成为研究关注的重点之一。近年来,基于实验光电子能谱(photoelectron spectroscopy,PES)和理论第一性原理计算,大量平面或准平面、管状硼环及双链硼纳米带结构的硼团簇被陆续发现[4-11]。随后,利用金属掺杂纯硼团簇产生了一系列新颖结构和特殊性质的B掺杂体系,并以此结构为基元,构建了众多功能和纳米新型材料[12-21]。例如:基于实验的PES和密度泛函理论的第一性原理计算,美国布朗大学的王来生教授课题组发现了一系列特殊结构的B掺杂体系,例如:RhB9-、IrB9-、RhB9、IrB9、CoB8-、RuB9-、TaB10-和NbB10-等团簇呈现掺杂原子位居中心的轮状平面结构[12-14],CoB12-和RhB12-的基态呈现半三明治夹心结构[15],AlB7-(C7v)和AlB8-(C8v)团簇的基态具有高对称性的伞状结构(Al原子位居伞的顶端)[16]。最近,又发现了高配位数CoB16-具有高对称性(能量几乎简并D8d和C4v)鼓状结构[17]。此外,其他课题组陆续发现了类平面(RhB18-)、双环状(LiB20、NaB20、KB20、CaB20、MgB20)、笼状(TiB22和 ScB22)及三环管状(Li2B24)等结构[18-21]。
具有空d电子轨道的V元素在地壳中具有很高的丰度,易于同其它材料结合形成新颖结构材料。近年来,众多科研工作者对过渡金属V原子掺杂硼团簇进行了深入研究[22-24]。Li课题组研究了VB10-团簇的结构及芳香性,结果发现VB10-基态具有船型结构[22]。Chen等研究发现VBn+(n=3~6)同甲烷反应的过程中,很容易使甲烷脱氢,该研究为甲烷的催化和转换指明了方向[23]。2019年,Pham课题组基于第一性原理计算,研究得知V2B70/V2B7+/V2B7-具有陀螺型结构[24]。基于此结构,对体系的磁性、芳香性及与二氧化碳分子的作用进行了深入探究。此外,基于量子化学计算,Tran等研究V2B70/V2B7+/V2B7-团簇的几何和电子结构[25],优化结构发现,中性和阳性体系的基态具有Cs点群对称的平面结构。此外,分析了V2B7+团簇同甲烷反应的机理。最近,我们课题组对过渡金属V掺杂硼团簇的结构和性质进行了一系列研究[26-28]。例如,结合密度泛函理论和卡里普索(crystal structure analysis by particle swarm optimization,CALYPSO)结构预测程序,优化得到了拥有高对称性(C2v点群对称)管状结构的VB16-[26];紧接着,在VB16-高对称结构基础上,通过堆积的方式获得了高对称V2B24-和V3B32-(C4h和D8d点群对称)团簇[27]。最近,基于相同的方法,系统搜索了VB20-团簇的几何结构,优化得到了高对称性管状分子马达[28]。在此结构基础上,研究了电子、光谱特性,揭示了其结构流变的本质。目前为止,对不同尺寸下V掺杂硼团簇几何结构、电子和热力学特性的研究未见报道。本文中,基于前期工作基础,利用卡里普索结构预测程序,并结合密度泛函理论的第一性原理计算,首先搜索了不同尺寸下VB2n-(n=8、9、10、11、12)团簇的基态和亚稳态结构。基于基态结构,分析了不同尺寸下的团簇的相对稳定性,研究了掺杂体系的电荷转移和极化率,拟合出了PES、红外和拉曼谱图,分析了流变键和芳香特性。最后讨论了体系的热力学特性和温度对热力学参数的影响。
1 计算方法
研究可知,合理基态结构的确定是研究其物理和化学特性的前提和基础。采用卡里普索结构预测程序对VB2n-(n=8~12)团簇的结构进行了系统搜索[29-31]。具体搜索过程如下:首先,基于不同自旋多重态(2S+1=1、3、5、7),在 PBE/3-21G水平下对初始结构进行搜索,根据设置的种群数(20个)和演化的代数(50代),可以得到相应数量的初始结构(总结构数=种群数×代数),从上述初始结构中筛选出能量差超过0.3 eV的50个结构。随后,在高精度的PBE0/6-311+G(d)水平下对筛选的50个结构进行重新优化[32-34],根据优化后能量大小关系,即可得到研究对象的基态和亚稳态结构。对于此预测程序的具体描述可以参考相关已发表的文献[26-28,35-47]。此外,为了考虑不同泛函对优化结果的影响,在相同的基组下,利用TPSSH泛函对50个能量差超过0.3 eV的结构进行了优化[48]。众多研究已证明TPSSH泛函可以应用到过渡金属原子掺杂硼团簇的研究[24,49-51]。所有优化过程中不设置对称性限制并考虑了自旋多重度的影响,上述运算均在Gaussian 09程序下运行[52]。
2 结果与讨论
2.1 VB2n-(n=8~12)团簇结构和稳定性
通过卡里普索结构预测程序,在PBE0/6-311+G(d)和TPSSH/6-311+G(d)水平下对纯硼B-和VB-2n2n(n=8~12)团簇的结构进行了搜索优化,优化后基态及亚稳态结构见图1。首先,优化得到的纯硼团簇的基态和美国布朗大学王来生教授课题组通过PES实验与理论研究所得的结果完全吻合[52],这充分证明所采用泛函基组的合理性。对于掺杂体系,本课题组已对VB16-和VB20-的基态结构做过详细报道[26,28]。其次,对于掺杂体系,2种泛函下的基态和亚稳态完全相同,说明优化的基态结构是合理的。最后,对比掺杂前后结构发现,V掺杂后完全改变了原团簇的结构,形成了具有高对称性的鼓状(VB16-)、管状(VB18-和VB20-)和笼式(VB22-和VB24-)结构,而且,掺杂的V原子都位居内部,每个团簇基态对应的点群对称性分别为C2v、C2v、Cs、C2和D3h。此外,图2给出了VB2n-(n=8~12)团簇的分子轨道能级,对应的HOMO-LUMO能隙分别为2.076、3.039、2.987、3.283和4.059 eV;而对应未掺杂体系B2n-(n=8~12)团簇的HOMO-LUMO能隙分别为1.671、1.493、1.578、1.542和1.544 eV,数据对比发现,掺杂后体系的HOMOLUMO能隙明显增大,说明V掺杂硼团簇VB2n-(n=8~12)的稳定性提高了。

图1 VB2n-(n=8~12)团簇的基态及亚稳态结构、点群对称、电子组态及PBE0/6-311+G(d)和TPSSH/6-311+G(d)水平下的相对能量Fig.1 Lowest energy and low-lying structures VB2n-(n=8-12)clusters along with the point group symmetry,electronic state,and relative energies at PBE0/6-311+G(d)and TPSSH/6-311+G(d)levels

图2 VB2n-(n=8~12)团簇基态结构的分子轨道能级Fig.2 Molecular orbital energy levels of the lowest energy structures of VB2n-(n=8-12)clusters
为了研究体系的稳定性,在PBE0和TPSSH泛函下计算了体系的平均束缚能(Eb):
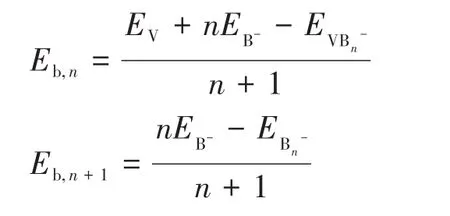
其中,E为对应原子和团簇的能量。图3给出了平均束缚能随尺寸变化的关系,从中可以看出,掺杂体系的平均束缚能大于纯硼团簇的平均束缚能,说明掺杂体系的稳定性强于对应的纯硼团簇的稳定性,该结论同HOMO-LUMO能隙分析结论完全相同。同时,表1给出了不同尺寸团簇中V—B和B—B键的Wiberg键级的大小,从中可以看出,每个掺杂体系中V—B键级大于B—B键级,说明V—B键比B—B键更稳定。
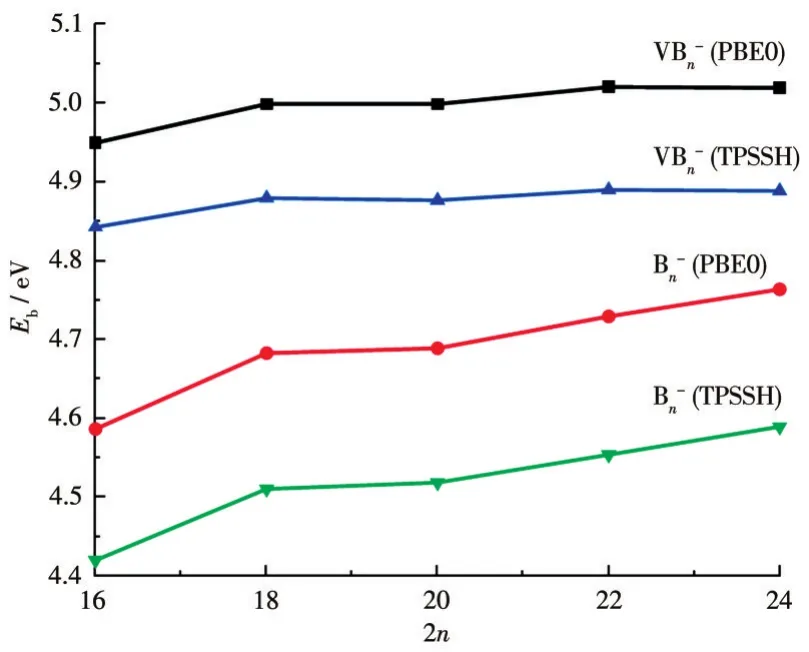
图3 PBE0和TPSSH泛函下VB2n-(n=8~12)团簇基态结构平均束缚能随尺寸变化Fig.3 Clusters size dependence on the binding energy of the lowest energy structures of VB2n-(n=8-12)clusters
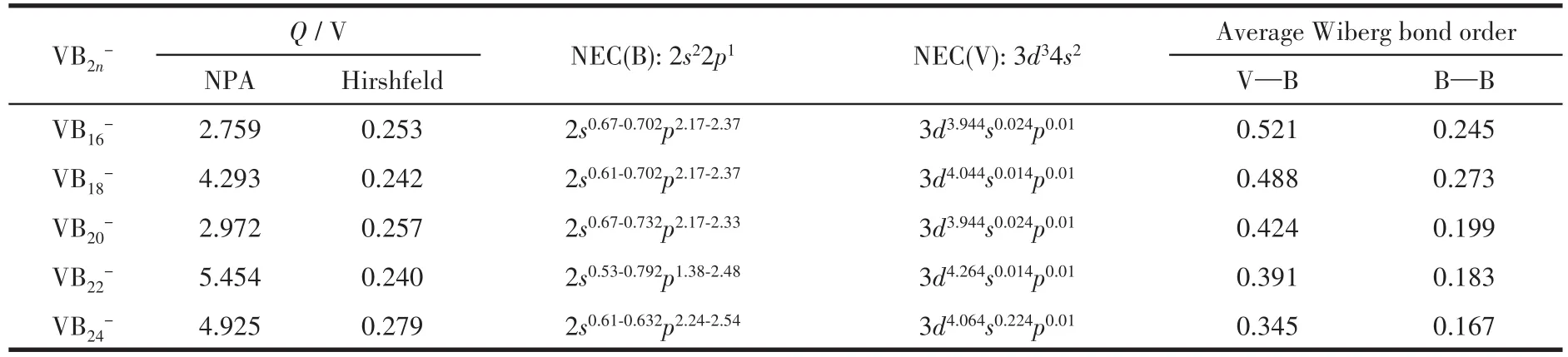
表1 VB2n-(n=8~12)团簇基态结构中V原子的净电荷(Q)、NEC、键长和Wiberg能级Table 1 Atomic net charges(Q)of V atoms,NEC,bond lengths,and Wiberg bond order of the lowest energy structures of VB2n-(n=8-12)clusters
2.2 电荷转移
为了了解团簇内部电荷转移信息,利用不同的电荷分析方法对电子在原子间及内层轨道上转移特征进行了分析,具体结果见表1。对比发现,基于自然布居分布(natural population analysis,NPA)和Hirshfeld分析方法得到的V原子的净电荷均为正值,说明在VB2n-(n=8~12)体系中,电子的转移方向为V→B,这与V的电负性(1.63 eV)小于B原子的电负性(2.04 eV)相吻合[53]。众所周知,自由B原子的电子排布为1s22s22p1,自由V原子的电子排布为[Ar]3d34s2。基于自然电子组态(natural electron configuration,NEC)方法计算发现,B原子的B2s轨道失去电子,B2p轨道得到电子。然而,V3d轨道得到电子,V4s轨道失去电子。数据说明,电子从B2s和V4s轨道转移到B2p和V3d轨道,B原子和掺杂的V原子之间存在强烈的spd轨道杂化。此外,自由V原子的价电子排布为3d34s2,当V原子掺杂硼团簇后,3d轨道得到大约1个电子,而4s轨道上的2个价电子几乎全部转移到其它轨道,即3d轨道得到一个电子,4s轨道失去2个电子,所以,VB2n-(n=8~12)体系中V均约为+1价。
2.3 极化率
为了考察VB2n-(n=8~12)对外场的响应,计算了各向同性平均极化率α、团簇平均极化率、各向异性极化率Δα,其中和Δα定义如下:

式中,N为团簇的总原子数,αxx、αxy、αxz、αyy、αyz和αzz为张量对角元,具体计算结果见表2。对比数据发现,VB16-团簇的平均极化率最大,说明VB16-团簇的电子结构相对稳定性差,容易被外加场极化,非线性光学效应强。VB24-团簇平均极化率最小,其相对稳定性强,最不容易被外加场极化,非线性光学效应弱。其次,VB16-团簇各向异性极化率最大,说明对外场的各向异性响应较强;VB24-团簇各向异性极化率最小,说明对外场的各向异性响应最弱。最后,对比HOMO-LUMO能隙、平均极化率和各向异性极化率发现,三者之间存在反比关系,较大的HOMO-LUMO能隙对应较小的平均极化率和各向异性极化率,原因可能是在微弱外电场下,HOMOLUMO能隙越大,电子跃迁时的阻力越大,因此,能转移的电子数越少,形成的平均极化率和各向异性极化率越小。
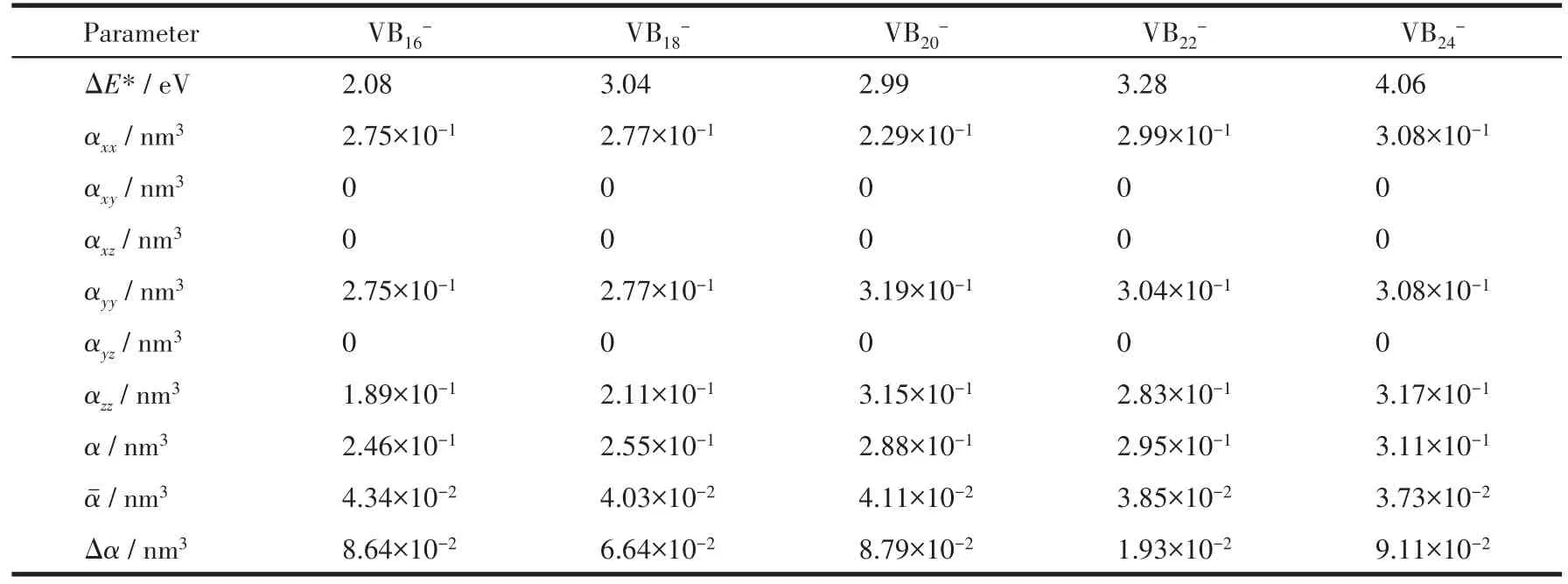
表2 VB2n-(n=8~12)团簇基态对应的HOMO-LUMO能隙和极化率Table 2 HOMO-LUMO energy gaps and polarizabilities of lowest energy structures of VB2n-(n=8-12)clusters
2.4 PES、红外、拉曼谱图
光电子技术是研究物质电子结构的重要方法,是鉴别其结构的一个重要手段。我们利用Multiwfn软件拟合出VB2n-(n=8~12)团簇的 PES(图4)[54],希望理论拟合的PES谱图为实验上鉴别VB2n-(n=8~12)团簇的基态结构提供有力的支持。
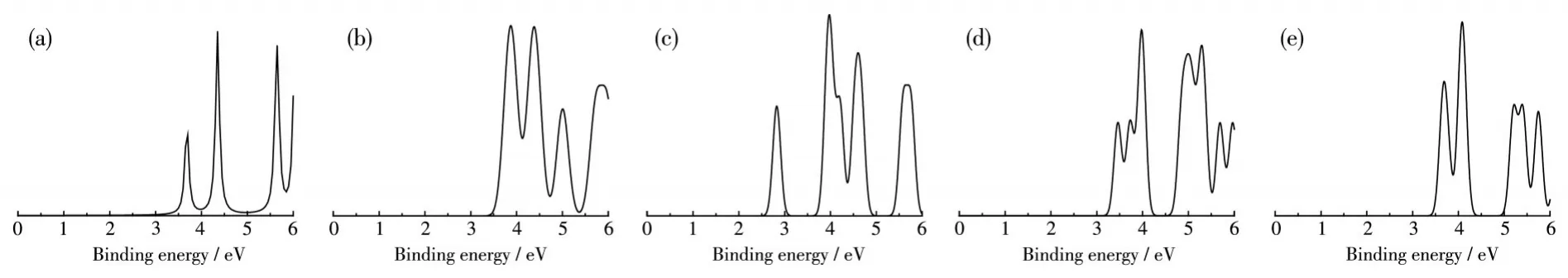
图4 (a)VB16-、(b)VB18-、(c)VB20-、(d)VB22-及(e)VB24-团簇基态结构的PES谱图Fig.4 PES spectra of the lowest energy structures for(a)VB16-,(b)VB18-,(c)VB20-,(d)VB22-,and(e)VB24-clusters
根据已知的基态结构,计算并拟合出VB2n-(n=8~12)团簇的红外和拉曼谱图(图5和6)。首先,红外和拉曼谱图的主要特征峰集中在200~1 400 cm-1范围内。对于鼓状结构VB16-,其红外谱图的主要特征峰位于461 cm-1处,表现为所有B5、B7、B11和B17的左右摇摆振动,而剩余的B原子和掺杂V原子受迫做水平振动。拉曼谱图的主要特征峰在715 cm-1处,表现为所有B原子围绕管中心V原子径向向里的呼吸振动模式,而中心的V原子几乎不动。VB-18的红外和拉曼谱图的主要特征峰在495和718 cm-1处,分别为B2单元中的B16和B17的左右摇摆振动和所有B原子径向向外的呼吸振动模式。对于VB20-,红外谱图的主要特征峰在386 cm-1处,表现为中心V原子沿Z轴平面方向的左右水平振动,而剩余的B原子做受迫振动。拉曼谱图的主要特征峰在665 cm-1处,所有B原子做径向向外的呼吸振动,其中,B11和B18沿着V—B键方向,上下环中的B原子沿着上下环的B—B键方向。VB22-的红外和拉曼谱图的主要特征峰分别在368和661 cm-1处,分别对应所有原子的左右摇摆振动(掺杂V原子沿Z轴方向运动)和所有B原子沿不同径向方向的呼吸振动模式。对于VB24-,红外谱图的主要特征峰在332 cm-1处,表现为中心V原子沿Z轴方向的左右水平振动模式,拉曼谱图的特征峰(624 cm-1)表现为所有B原子沿B—V径向方向的呼吸振动模式。

图5 (a)VB16-、(b)V B18-、(c)VB20-、(d)VB22-及(e)VB24-团簇基态结构的红外光谱和最强峰的振动模式图(插图)Fig.5 IR spectra and vibrational mode of the strongest peak(Inset)for(a)VB16-,(b)VB18-,(c)VB20-,(d)VB22-,and(e)VB24-clusters
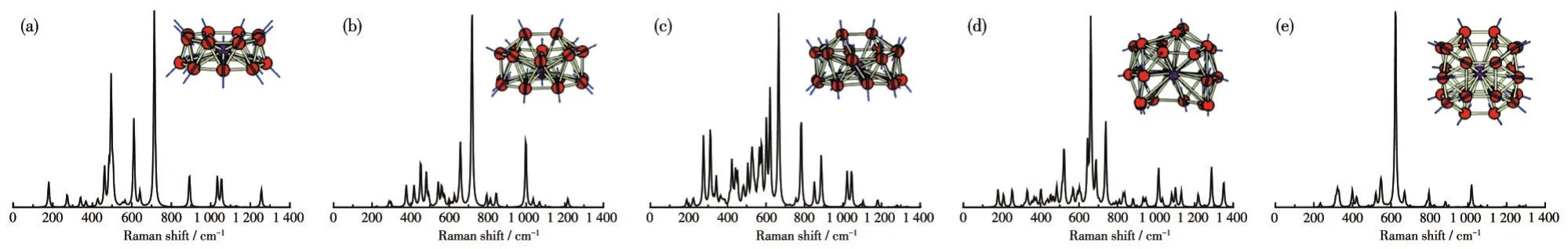
图6 (a)VB16-、(b)VB18-、(c)VB20-、(d)VB22-及(e)VB24-团簇基态结构的拉曼光谱和最强峰的振动模式图(插图)Fig.6 Raman spectra and vibrational mode of the strongest peak(Inset)for(a)VB16-,(b)VB18-,(c)VB20-,(d)VB22-,and(e)VB24-clusters
2.5 流变键
流变键(fluxional bonds)是由山西大学李思殿教授课题组首次提出[55-57],用以描述一定条件下通过协同机制不断断裂和生成的化学键。基于第一性原理计算和适应性自然密度划分(adaptive natural density partitioning,AdNDP)法,该课题组对准平面结构B19-、管状 TaB20-、硼球烯 B39-[55]、半三明治结构MB18-(M=K、Rb、Cs)[56]及 NiB11-[57]进行了详细的流变键分析。其中,研究管状TaB20-发现,上方的 B2单元与管底座[TaB18]间形成的2个σ键在一定温度下均不断地断裂和生成,致使该团簇在基态和过渡态之间发生流变。鉴于VB20-和TaB20-团簇具有相同的价电子数(33对),以VB20-团簇为例,通过对基态和过渡态进行结构的流变和AdNDP键合分析,研究了其流变的机理。对于基态结构,管上方B2单元的B—B键长为0.159 7 nm,比与管连接的B—B键长(0.171 5和0.187 3 nm)短,说明B2单元中B—B键能大于B2单元和管连接的B—B键能,当发生流变时,B2单元和管连接的B—B易断裂。首先,通过优化的基态结构确定了其过渡态结构,图7为VB20-团簇在势能面上的结构流变图。从中可以看出,对于基态,管上方的B2单元与管顶部B9环之间有2个4c-2eσ键。当上方的B2单元克服8.46 kJ·mol-1的流变能垒后,可以绕底座[VB18]开始顺时针旋转,进而流变到过渡态,此时2个4c-2eσ键就变成了2个3c-2eσ键。因此,VB20-团簇的基态与过渡态之间发生流变。
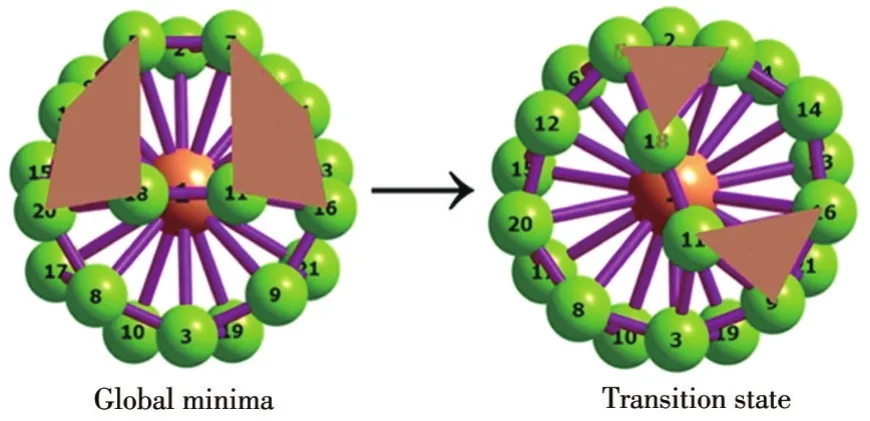
图7 VB20-团簇基态的结构流变键图Fig.7 Fluxional bonds of the lowest energy structure of VB20-cluster
为了深入研究其流变机理,对VB20-的基态和过渡态进行了AdNDP分析(图8)。其中,nc-2e代表n中心双电子键,红色和蓝色代表不同的相。为了方便对比VB20-的基态和过渡态中流变键的变化,图8仅提供了前22对价电子数的AdNDP。对于基态和过渡态,首先,位于[VB18]管上端的B—B形成了1个2c-2e的双中心双电子σ键,占据数为1.82e和1.81e。其次,管上方B2单元和中心V原子之间形成一个3c-2eσ键,对应的占据数为1.97e和1.96e。对于B18的管状而言,每相邻的3个B原子之间构成了18个3c-2eσ键,其占据数分别为1.91e~1.93e和1.90e~1.96e。对于过渡态,管顶部B9中的B原子和B2单元B原子之间构成了2个3c-2eσ键;对于基态,继续搜索可以得到管顶部B9中的B原子和B2单元B原子之间构成了2个4c-2eσ键,基态和过渡态中的占据数分别为1.79e和1.88e。总之,二者的不同之处在于,在基态中是2个4c-2eσ键,而在过渡态中是2个3c-2eσ键,该分析同上文中对流变键的分析相符。

图8 VB20-团簇AdNDP成键图Fig.8 AdNDP bonding patterns of VB20-cluster
2.6 芳香性
核独立化学位移(nucleus independent chemical shifts,NICS)方法是一种分子芳香性的判断依据,广泛应用于有机无机化合物及团簇芳香性的研究中,NICS为负值表示芳香性,NICS为正值表示反芳香性,当NICS值接近零时为非芳香性。本工作中,计算NICS时,管状和笼状结构的参考点分别为管状和笼状结构的中心,试探原子Bq分别在其中心的0.05、0.10、0.15和0.20 nm处,具体计算结果见表3。结果发现,VB20-和VB24-具有芳香性。

表3 VB2n-(n=8~12)团簇基态结构的NICS值Table 3 NICS values of the lowest energy structures of VB2n-(n=8-12)clusters
2.7 热力学性质
在298.15 K、1.013×105Pa下,计算了VB2n-(n=8~12)团簇的热力学参数:定容热容Cv、标准熵S和标准生成焓H,其中H可表示如下:

计算结果见表4,从中可以看出,所有体系的生成焓均为负值,说明生成团簇的反应都是放热反应,热力学上都是稳定的,而且,随着团簇尺寸的增大,稳定性逐渐提高。为了探究温度对Cv和S的影响,计算了不同温度下的Cv和S值,具体结果见表5和图9。结果发现,热力学参数Cv和温度之间存在二次函数关系,S和温度之间存在近似线性关系。
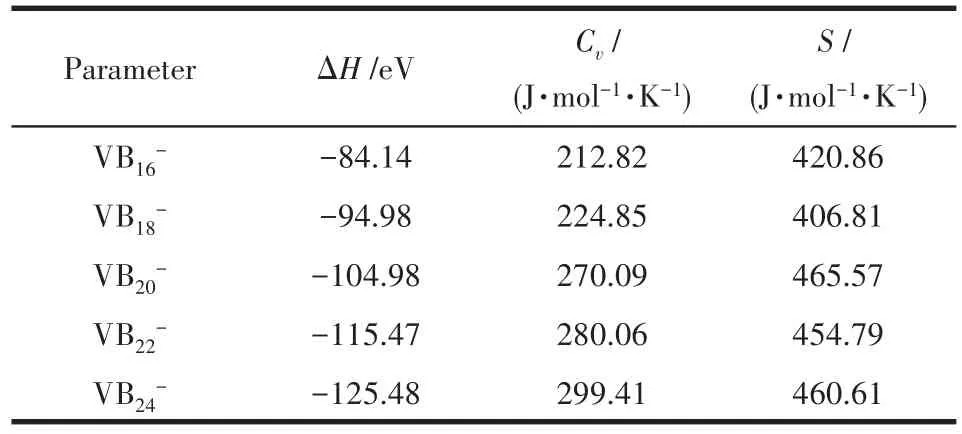
表4 计算得到的VB2n-(n=8~12)团簇基态结构热力学参数Table 4 Calculated thermodynamics parameters of the lowest energy structure of VB-2n(n=8-12)clusters
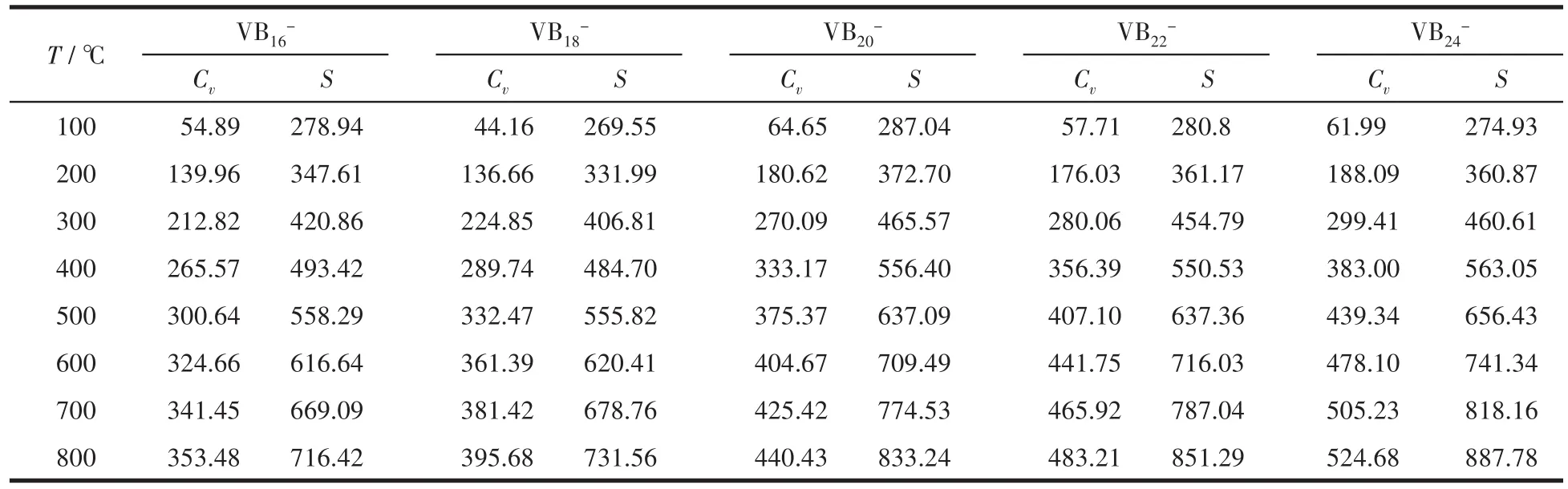
表5 不同温度下的定容热容和标准熵Table 5 Constant heat capacities and standard entropies at different temperatures J·mol-1·K-1
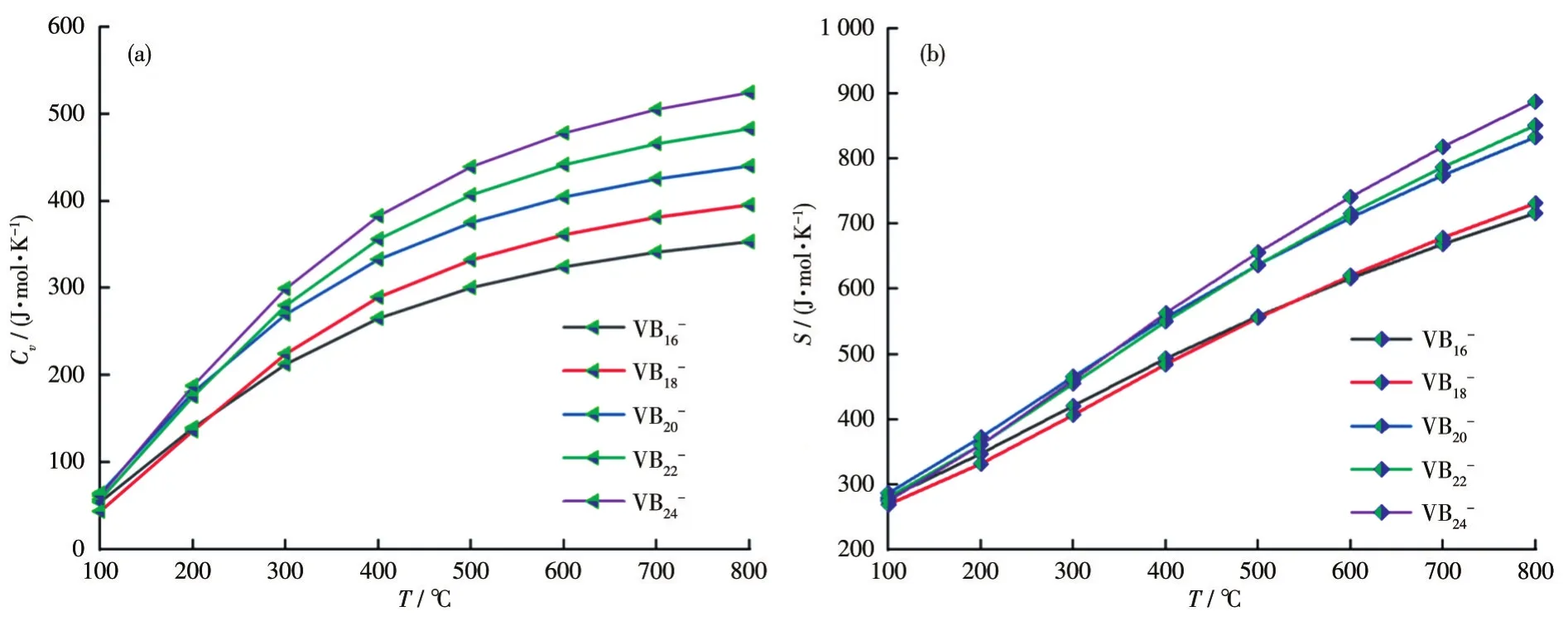
图9 VB2n-(n=8~12)团簇的(a)Cv和(b)S随温度变化的曲线Fig.9 Temperature dependence curves of(a)Cvand(b)S for VB2n-(n=8-12)clusters
3 结论
结合密度泛函理论和卡里普索结构预测程序,系统研究了过渡金属V原子掺杂硼的VB2n-(n=8~12)团簇的几何结构、稳定性、电子和热力学特性。结构优化发现,V的掺杂完全改变了纯硼团簇的结构,掺杂体系的基态结构分别呈现鼓状(VB16-:C2v)、管状(VB18-:C2v和VB20-:Cs)及笼状(VB22-:C2和 VB24-:D3h)构型,掺杂提高了硼团簇的稳定性。电荷转移分析发现,在原子之间,电荷从V原子转移到硼原子;在原子内部轨道上,电子从B2s和V4s轨道向B2p和V3d轨道转移,B和V原子之间存在强烈的spd轨道杂化。极化率分析发现,易被外加场极化的VB16-拥有最大的平均极化率和各向异性极化率,不容易被外加场极化的VB24-拥有最小的平均极化率和各向异性极化率。而且,HOMO-LUMO能隙、平均极化率和各向异性极化率之间存在反比关系。拟合得到的光电子能谱、红外和拉曼谱图有望为以后的实验研究提供帮助。最后,对管状VB20-的成键分析发现,基态结构上方B2单元分别与管顶部的3个B原子之间形成2个4c-2eσ键;过渡态结构上方B2单元分别与管顶部的2个B原子之间形成2个3c-2eσ键,基态与过渡态之间发生流变。此外,芳香性研究发现,VB20-和VB24-具有芳香性。热力学性质分析表明,所有研究体系在热力学上都是稳定的,而且定容热容和温度之间存在二次函数关系,标准熵和温度之间存在近似线性关系。
猜你喜欢
现代财经-天津财经大学学报(2022年5期)2022-06-01
航天电子对抗(2022年2期)2022-05-24
中国药学药品知识仓库(2022年8期)2022-05-09
电子乐园·上旬刊(2022年5期)2022-04-09
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
北京航空航天大学学报(2021年9期)2021-11-02
宝藏(2021年11期)2021-01-01
航天电子对抗(2019年4期)2019-06-02
